氮化碳基復合材料的研究進展
2023-09-21 05:10:02肖舒寧
有色金屬材料與工程 2023年4期
關鍵詞:復合材料
肖舒寧, 黃 倩
(上海理工大學 材料與化學學院,上海 200093)
當前,光催化技術的可持續發展,使其被廣泛認為是解決全球能源和環境問題的最重要手段之一。受植物光合作用的啟發,研究人員一直在努力開發可在綠色、環保和可持續條件下進行的新有機反應[1]。
聚合物石墨相氮化碳(graphitic phase carbon nitride, g-C3N4)是一種僅含有地球上豐富的碳和氮元素的高活性光催化劑[2]。在光催化產氫、光還原CO2、以及有機物的降解等領域,g-C3N4納米復合材料得到了廣泛探索[1,3-6]。可見光光催化是有機轉化的一個強大的新興領域,具有很高的可觀性[7],與g-C3N4的結合能夠進行有機化合物的功能化[8-10]。然而,關于g-C3N4基復合材料的制備以及改性后光催化性能的比較是非常有限的。本文綜述了3 種不同的g-C3N4的合成方法,詳細討論了各種方法在制備g-C3N4方面的優點。此外,還闡述了改善g-C3N4光催化活性的改性方法,并對其的應用與發展進行了分析和展望。
1 半導體光催化機制
光催化是一種化學反應,能觸發或加速與輻照半導體的特定還原和氧化(氧化還原)反應[11]。在半導體中,與標準氫電極(normal hydrogen electrode,NHE)相比,當導帶(conduction band, CB)電子與普通氫電極相比具有?1.5~0.5 V 的化學勢時,光催化劑可以用作還原劑;與NHE 化學勢相比,在價帶(valence band, VB)的空穴具有1.0~3.5 V 的化學勢時,光催化劑表現出很強的氧化性。一旦入射光子的能量匹配或超過光催化劑的帶隙,光吸收立即進行,隨后電子–空穴對的光激發過程主要包括3 個關鍵步驟[12]:(1)太陽光照射引起的電子–空穴對產生;(2)光激發的電子和空穴轉移到光觸媒表面;(3)還原/氧化反應發生在光觸媒表面。綜上所述,影響光催化反應速率的因素主要包括:(1)光催化劑的光吸收范圍;(2)光生電荷的轉移和分離效率;(3)光催化劑表面的催化反應速率;(4)光生載流子的氧化還原能力。
2 氮化碳基半導體光催化劑的合成
2.1 簡單的煅燒法
調控熱化學反應環境,可用于通過熱分解和聚合含氮和碳材料的前驅體制備g-C3N4。而在坩堝中使用g-C3N4前驅體或加入其他催化材料組分,并在必要的溫度下煅燒,可產生基于g-C3N4的納米復合材料。由于簡單、有效,該工藝被廣泛用于制造基于g-C3N4的復合材料。結果表明, 所形成的納米復合材料的光電特性得到了很大的改善。例如,Ismael 等[13]使用簡單的煅燒工藝開發了鈣鈦礦鐵酸鑭(perovskite, LaFeO3)/g-C3N4雜化復合材料。在另一項工作中,Ismael 等[11]使用簡單的煅燒方法來開發鈣鈦礦鐵酸釔(perovskite, YFeO3)/g-C3N4復合物。此外,Geng 等[9]通過高溫煅燒尿素獲得了g-C3N4納米片,并與Fe2O3復合得到Z 型異質結。
2.2 水熱法
水熱合成方法不同于其他常規方法,具有降低反應溫度、單晶生長和調節顆粒形貌等特點。Mao等[8]報道了通過水熱法從1.3.5-三氯三嗪合成了g-C3N4,并使用AgNO3和NaHCO3溶液將Ag2CO3納米顆粒沉積到其表面。Ag+通過化學吸附與表面結合,生成Ag(NH3)2+和CO32?,離子交換反應形成g-C3N4/Ag2CO3復合材料。Feng 等[14]通過水熱制備質子化的g-C3N4,設計了AgBr/g-C3N4納米復合材料。然后使用十六烷基三甲基溴化銨(cetyl trimethyl ammonium bromide, CTAB)和AgNO3的水溶液摻入AgBr 顆粒。質子化效應導致g-C3N4表面缺陷數量的增加,從而為多孔AgBr/g-C3N4納米復合材料中的AgBr 晶粒提供了生長位點。Vadivel等[15]通過水熱法合成了Ag-BiOF/g-C3N4復合材料。在該方法中,Ag 納米粒子和g-C3N4分散在BiOF方塊表面。Zhang 等[16]通過對飽和NH4X(X=Cl,Br)溶液的水熱處理制備了對H2O2生成具有優異光催化活性的鹵素摻雜的g-C3N4納米棒。飽和鹽溶液水熱處理改變了所制備的g-C3N4催化劑的形貌和比表面積,并在g-C3N4晶格中摻雜了鹵素原子。Br 摻雜的g-C3N4催化劑表現出比Cl 摻雜的g-C3N4催化劑更高的光催化H2O2生產能力。
2.3 微波法
g-C3N4通常是通過將富 N 前驅體置于高溫下而獲得,這既費時又費力。受微波(microwave, MW)輔助制備石墨烯方法的啟示,Yuan 等[17]報道了g-C3N4的快速合成,即MW 輔助加熱方法快速構建復合材料。如圖1(a)所示使用MW 輔助加熱策略,g-C3N4光催化劑可在很短時間內獲得。有趣的是,通過這種方法制備的g-C3N4表現出優異的光催化產氫活性。這種改進的光催化性能可能是由于g-C3N4的結晶質量提高,導致比表面積增大以及光生電子和空穴的有效分離。為了在MW 輻射下獲得穩健可靠的合成條件,應優先考慮液體反應系統,因為MW 輻射在液體介質中比在常見的固態反應系統中更穩定、更高效。因此,水溶液、多元醇和離子液體在MW 輔助反應中被廣泛用作反應介質,以提高MW 輻射的穩定性。受此想法的啟發,Liu等[18]開發了一種MW 輔助熔鹽策略,使用易于獲取的三聚氰胺作為前驅體,制備基于三嗪/庚嗪的g-C3N4光催化劑的同型異質結。如圖1(b)~(d)所示,這種合成策略實現了g-C3N4光催化劑的大量且快速的生產和制備。MW 加熱和熔融鹽液體縮聚的協同效應為大規模和快速制備g-C3N4基光催化劑提供了強大和可靠的平臺。受此理念啟發,將多種方法結合起來實現高催化活性g-C3N4基光催化劑的規模化生產是非常重要的。
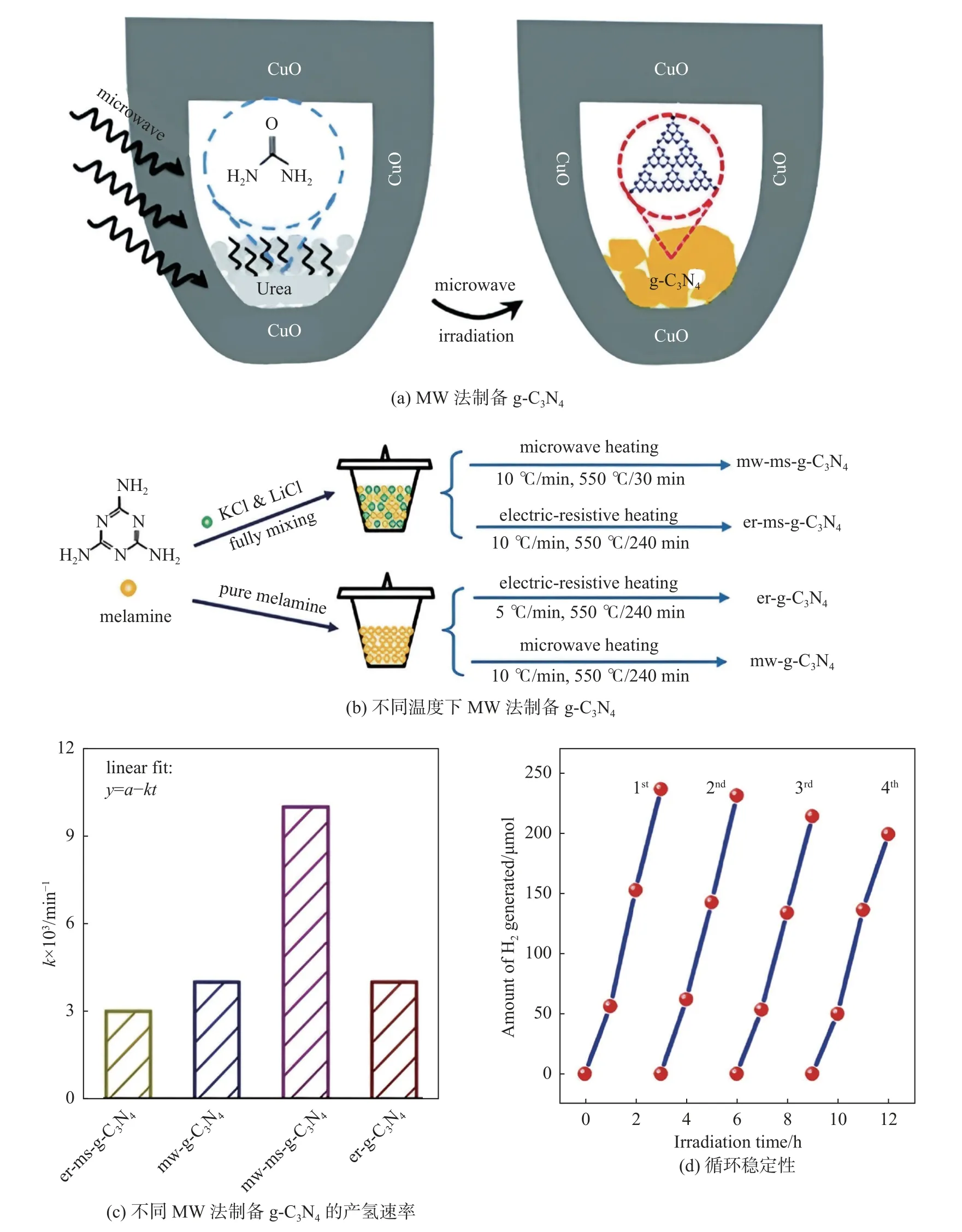
圖1 MW 法制備g-C3N4[17-18]Fig.1 g-C3N4 prepared by MW method[17-18]
3 氮化碳基光催化劑的改性策略
3.1 形貌調控
光催化劑活性取決于光催化劑的尺寸、表面形貌和結構。形貌調控是控制g-C3N4光催化活性的一種簡單且有效的方法。由于光催化劑具有更大的比表面積和更多的反應活性位點,可以實現光的多次反射和散射效應,從而達到更好的光催化效果[19]。例如, Yan 等[20]通過使用聚環氧乙烷(polyethylene oxide, PEO)嵌段聚合物作為更環保的軟模版,制備了窄孔徑分布的g-C3N4。獲得的介孔g-C3N4具有較大的比表面積和高達800 nm 顯著的紅移光吸收邊緣,以及在可見光下增強了光催化H2釋放速率。設計二維g-C3N4納米片和不同的多孔結構是應用最廣泛的形貌調控方法。通常,g-C3N4納米片具有更大的表面積、更薄的厚度、更高的載流子遷移速率以及固有的半導體特性。例如,Lu 等[21]使用一種簡便的一步雙氰胺發泡法,以NH4Cl 為氣體模板,實現了g-C3N4納米片的可擴展生產。一些獨特的三維結構,如納米棒網絡,多孔分級結構,為提高催化活性提供了強有力的支持[22]。例如,Wang 等[23]通過在PEO 軟模版的幫助下對超分子前驅體進行水熱處理,然后對獲得的中間體進行退火,制備了由空心氣泡組成的三維g-C3N4聚集體。三嗪分子的超分子化學制備具有多種形貌的氫鍵超分子網絡前驅體,可解決空心g-C3N4結構制備中的坍塌問題。結果表明,所制備的催化劑具有更大的比表面積、更高的可見光吸收活性和更多的反應活性位點。入射光可以在中空結構中連續反射,從而提高載流子產生效率,進而提高光催化活性。
3.2 元素摻雜
g-C3N4中庚嗪的存在會影響其電子結構。同時,g-C3N4基質的光學帶隙可以通過包含不同的元素摻雜劑以及碳和氧空位來減小,以增加要吸收的光的范圍。摻雜雜原子可以減小帶隙,同時還可以改變帶邊的電勢以提高g-C3N4的光氧化還原能力。Wang 等[24]通過密度泛函理論(density functionaltheory, DFT)計算研究了用于光催化CO2還原的S 摻雜g-C3N4。結果表明,S 摻雜g-C3N4顯示出帶隙變窄、光生載流子分離增強和壽命延長,有利于光催化CO2還原。Fu 等[25]通過連續熱氧化剝離從g-C3N4卷曲過程中構建了具有分級多孔結構的O 摻雜g-C3N4納米管,由于O 摻雜具有窄帶隙和快速電荷載流子分離的效果,其可見光光催化CO2還原活性提高了近5 倍(見圖2)。Cao 等[26]通過一步熱冷凝法可制備超薄S 摻雜g-C3N4納米片(S doped g-C3N4nanosheets,SCNNSs),其碳空位來源于自生成NH3氣氛中的硫脲分解。S 原子摻雜到g-C3N4中取代了晶格N 原子,優化后的光催化劑表現出優異的光催化固氮率,是原始的g-C3N4的2.8 倍。最近,Thaweesak 等[27]報道了使用雙氰胺作為g-C3N4前驅體、NH4Cl 作為剝離劑和三氫硼酸銨(H3NBH3)作為摻雜劑,通過一鍋熱縮聚法設計了一種新型黃色海綿狀B 摻雜的g-C3N4納米片。在B 摻雜后,g-C3N4的原始的堆疊多層結構也得以保持。然而,層狀結構在剝離后變薄,通過設計形貌,B 摻雜的g-C3N4納米片提供了更多的表面活性位點。該光催化劑在可見光下顯示出顯著增強的H2釋放活性(1880 μmol/(h·g)),是純g-C3N4的12 倍,以及良好的穩定性。
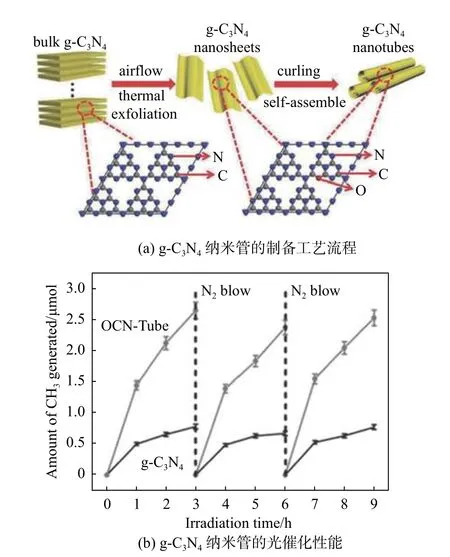
圖2 g-C3N4 納米管的制備及催化活性[25]Fig.2 Preparation and catalytic activity of g-C3N4 nanotubes [25]
3.3 助催化劑負載
助催化劑的負載對于光生載流子的提取和電荷分離效率的提高至關重要。在典型的g-C3N4/助催化劑體系中,g-C3N4是產生電荷載流子的光捕獲組分,而助催化劑促進電荷動力學并通過表面收集的俘獲電荷進行氧化還原反應。基于改進的電荷轉移和表面轉化效率,可以實現顯著增強g-C3N4的光催化活性。例如,以三聚氰胺為前驅體,通過簡單的煅燒制備的g-C3N4的析氫速率僅為0.13 μmol/(h·g)。相反,當原位光沉積的Pt 負載在g-C3N4上作為助催化劑,產氫速率可以提高到40 μmol/(h·g)左右,表明負載型助催化劑的顯著效果。Cao 等[28]成功制備了具有各種形狀的暴露面的負載Pt 助催化劑的g-C3N4,其光捕獲和電荷分離效率相當,但析氫性能不同。圖3 為不同CN 的SEM 圖及產氫速率圖。析氫差距的主要原因可歸因于不同暴露面的不同表面原子結構。球形Pt 可以暴露更多的Pt 的活性催化位點,這更有利于H2O 的吸附,從而實現最高的助催化性能。
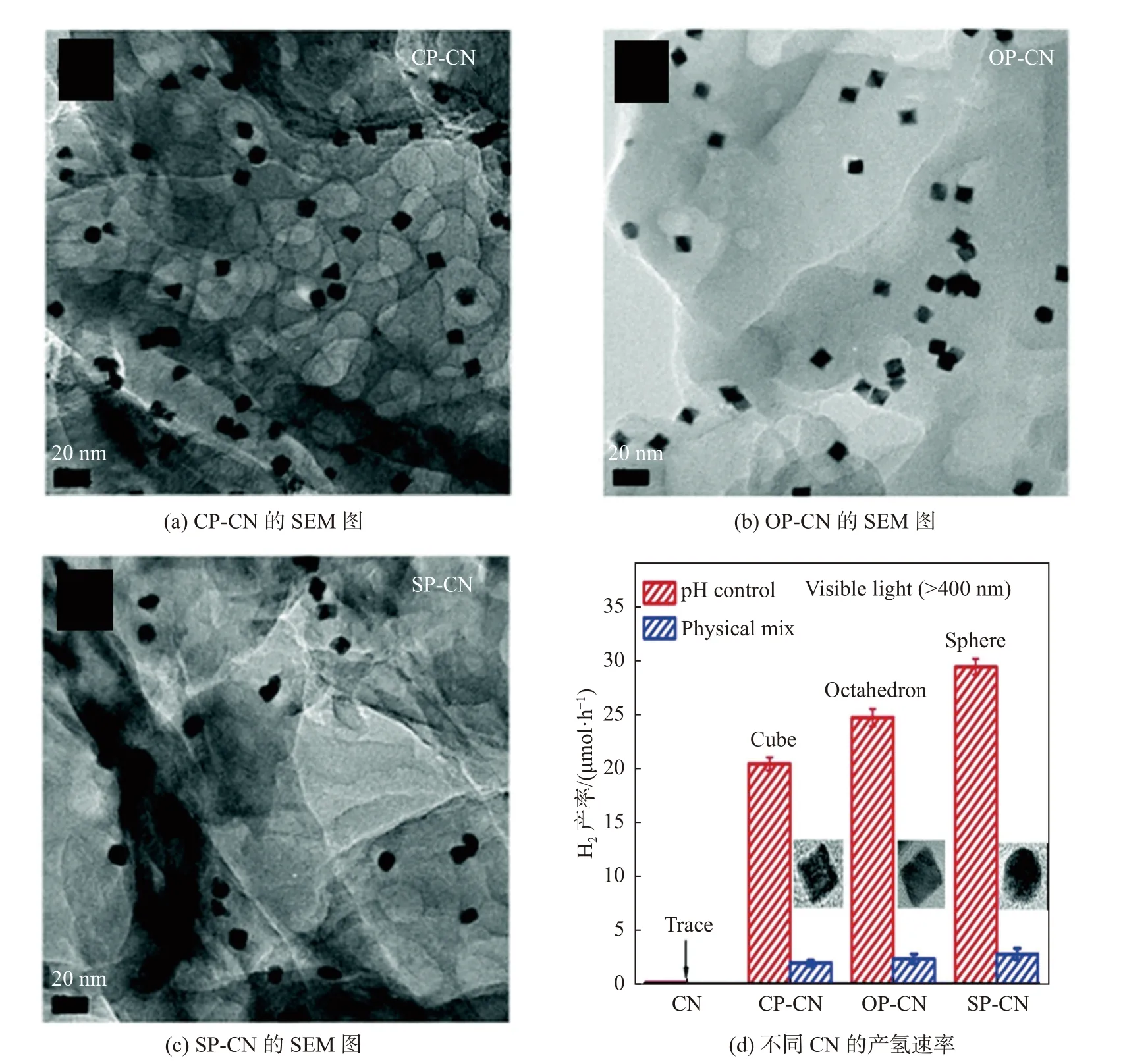
圖3 不同CN 的SEM 圖及產氫速率[28]Fig.3 SEM images of different CN and hydrogen production rates[28]
3.4 構建異質結
為了開發高效的g-C3N4基納米復合材料,光催化劑應具有合適的結構,比如窄帶隙能量以吸收太陽光譜中的可見光,以及用于高效氧化還原反應的合適的能帶位置。單個半導體無法滿足這些要求,而在兩種或多種半導體之間構建納米復合材料可以解決這些問題[7]。通過構建g-C3N4基異質結可以改善電荷載流子分離,從而提高g-C3N4基光催化劑的光催化活性。
傳統的g-C3N4II 型異質結是通過利用g-C3N4和另一種半導體結合而組建的,其中g-C3N4的CB 和VB 位置都比另一種半導體高或低。如圖4(a)所示,兩種半導體之間的化學勢差導致異質結接觸界面處的能帶彎曲。這種能帶彎曲將會產生內建電場,該電場可以驅動光生電子和空穴的反向遷移。特別地,當異質結被能量大于或等于兩個半導體的帶隙的能量的光子照射時,異質結中的兩個半導體可以被同時激發。兩個半導體之間的大接觸比表面積有利于跨異質結界面的有效空間電荷再分布。這種電荷重新分布可以大大提高光催化性能。在這方面,Zhang 等[29]通過表面輔助聚合獲得的傳統g-C3N4和S 摻雜g-C3N4同型異質結,形成比機械混合更多的連接界面。由于傳統g-C3N4的CB 和VB 位置均高于S 摻雜g-C3N4,形成了具有有效電荷分離的II 型異質結,從而對析氫具有更好的光催化活性。
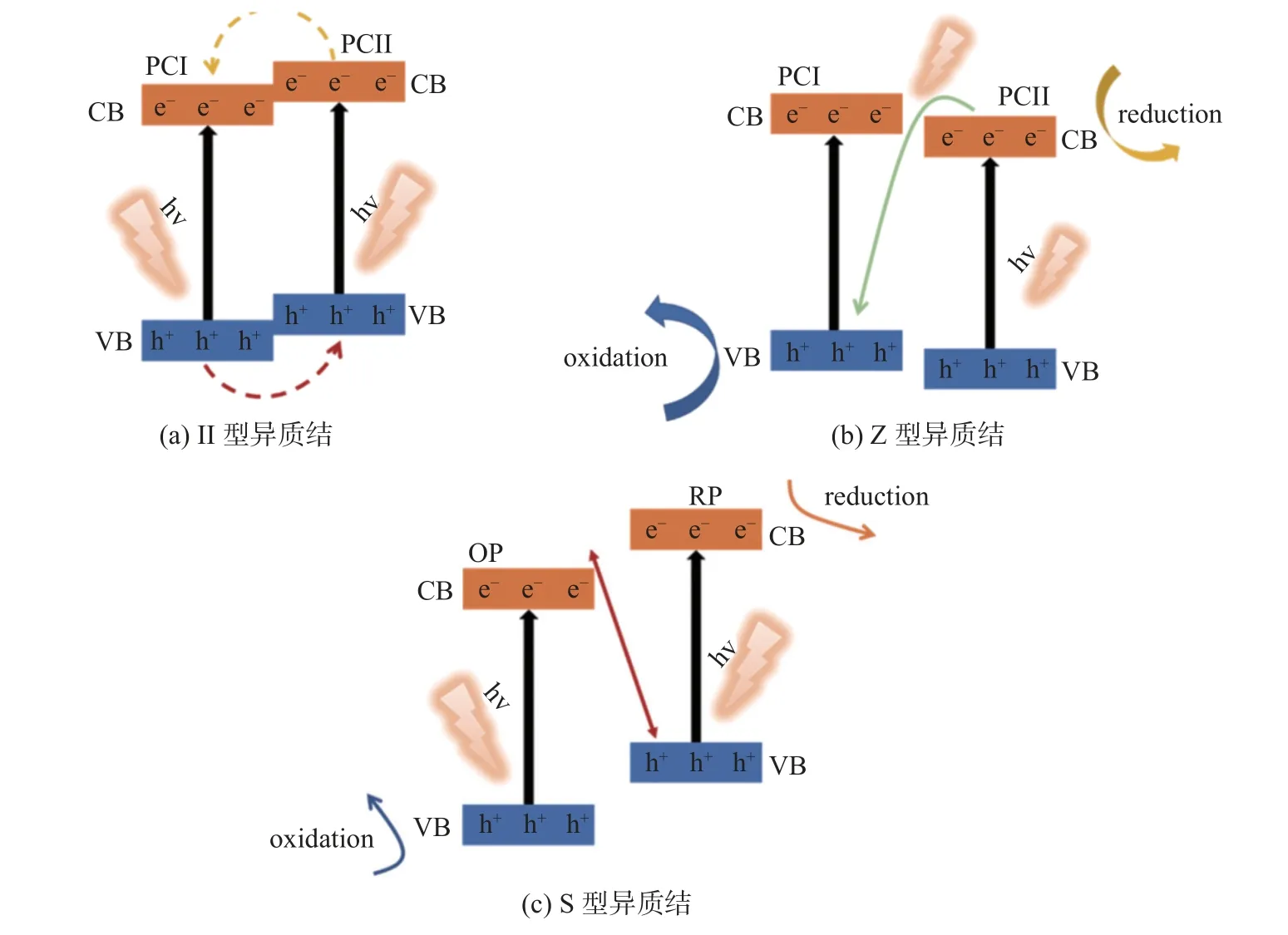
圖4 3 種異質結光催化機制Fig.4 Mechanisms of three heterojunction photocatalysts
傳統的II 型異質結雖然可以有效地實現空間電荷分離,但這種異質結的主要缺點是半導體II(photosemiconductor II, PC II)的CB 和VB 的正負性極低,光生電子和空穴的氧化還原能力較弱。在這種情況下,窄帶隙半導體對很難同時實現優異的電荷分離效率和強氧化還原能力,而Z 型異質結可以有效地解決這一問題。Z 型異質結與傳統的II 型異質結有很大不同。這樣的異質結可以利用具有窄帶隙的半導體對而不損失光生電子和空穴的強氧化還原能力,如圖4(b)所示。Kumar 等[30]報道了利用分散–蒸發法合成N 摻雜ZnO/g-C3N4雜化核殼納米片。通過研究羅丹明B (Rhodamine B, RhB)在N 摻雜ZnO/g-C3N4核殼結構下光催化降解的反應物質,提出了一種Z 型異質結機制。類似的Z 型異質結,如g-C3N4-WO3[31]、g-C3N4-MoO3[32]和g-C3N4-BiOCl2[33]也被報道。
最近Xu 等[12]提出了一種新的基于Z 型異質結機制的S 型異質結方案。如圖4(c)所示,這種S 型異質結由氧化型光催化劑(oxidized photocatalyst,OP)和還原型光催化劑(oxidized photocatalyst, RP)組成。當RP 和OP 接觸時,RP 中費米能級較高的電子會在界面處漂移到費米能級較低的OP。因此,RP 側失去電子并帶正電,相反,OP 側接受電子并帶負電。在光照射下,它驅動光生電子從OP 的CB 轉移到RP 的VB。此外,OP 中電子與RP 和能帶彎曲中的空穴之間的庫侖吸引力也有利于這種電荷轉移。S 型異質結體系共同實現電荷分離,表現出較強的光氧化還原能力,從而提高了光催化性能。例如,Guru 等[34]通過簡單的共沉淀方法報道了一種由支持Bi2O2CO3和NiFe-LDH 的g-C3N4納米片組成的新型S 型異質結。界面電子轉移和能帶彎曲的結合表明S 型電荷轉移機制, 從而提高了光電化學析氫和氧的效率。Yang 等[35]利用靜電自組裝和熱處理策略,在單層 Ti3C2Tx(二維材料,Mxene)轉化的 TiO2/C 上制備了由超薄g-C3N4納米片組成的S型異質結光催化劑。帶正電荷的質子化g-C3N4和帶負電荷的Ti3C2Tx 之間的范德華力使界面接觸緊密。由于g-C3N4和 TiO2/C 之間的大接觸面積,所制備的g-C3N4/TiO2/C 顯示出優異的光催化CO2還原效率,這非常有利于光生電荷的 S 型異質結的界面轉移。
4 石墨相氮化碳基光催化劑的應用
4.1 可見光催化產氫
與其他碳氫化合物燃料相比,氫氣具有更高的比能量[3,5,26,36-38],預計60 年后將成為主要的能量來源。因此,技術發展應該為制氫過程、輸送、儲存、轉化和應用開辟有效途徑[26,39-42]。目前,廉價且環保的氫氣生產技術是非常具有吸引力的研究領域。自1972 年Fujishima 等[43]的開創性工作以來,光催化水分解制氫已經取得了顯著進展。不含金屬的g-C3N4是一種很有前途的光催化材料。最近,Yu等[44]采用簡單沉淀法制備了Ni(OH)2/g-C3N4光催化劑用于光催化制氫。研究發現Ni(OH)2負載物質的質量分數為0.5%,產氫效率高達7.6 μmol/h。此外,Wu 等[45]通過水熱法合成了Ag2O/g-C3N4。發現Ag2O/g-C3N4納米復合材料在可見光照射下的析氫活性。該光催化劑顯示出高效的光催化析氫活性。Wang 等[24]在硼酸存在下合成 Pt/ g-C3N4用于太陽能燃料生產。他們的發現表明,硼酸的存在改善了 Pt 納米顆粒在g-C3N4上的分布,從而進一步改善了電荷分離。同樣的,Zhang 等[46]采用自組裝浸漬輔助聚合法成功合成了Ni-P 共摻雜的g-C3N4多孔納米管,該納米管表現出240.91 μmol/(h·g)的優異的光催化析氫速率。
4.2 可見光催化CO2 還原
在自然界綠色植物光合作用的啟發之下,光催化將CO2還原成增值化學品被認為是活的可再生能源和消除溫室效應的可行策略。然而,CO2還原是一個復雜而困難的反應。具有路易斯堿性功能的g-C3N4具有親核性質,有利于CO2的活化,被證明是應用于CO2還原有效光催化劑之一。Yang等[37]成功制備了兩種不同的g-C3N4光催化劑,塊狀和片狀g-C3N4,帶隙分別為2.77 eV 和2.97 eV。在紫外–可見光下,片狀g-C3N4對CH4表現出高的選擇性,而塊狀g-C3N4更傾向于生成CH3CHO。質子化多孔g-C3N4納米片暴露出更多的活性催化位點,其中CO2吸附達到6.6 cm3/g。Crake 等[47]采用水熱原位生長法合成了TiO2和g-C3N4組成的用于CO2還原的復合光催化劑,TiO2納米粒子在 g-C3N4納米片上有效生長。測量了復合材料對CO2的吸附能力,并測試了它們在光照下以H2作為還原劑氣固相體系中將CO2捕獲和轉化結合為一步過程的CO2光還原。在不添加任何貴金屬助催化劑的情況下進行催化測試。與其組成材料相比,該復合材料表現出增強的CO2吸附能力和光催化CO2轉化率(增加10 倍以上)。Cao 等[26]設計了在構建配體中具有B-H 鍵的硼咪唑骨架-20(簡寫為“BIF-20”)。與g-C3N4納米片相比,BIF-20/g-C3N4納米片復合材料顯示出強的光催化CO2還原活性。盡管g-C3N4納米片表現出優異的光催化性能,但效率仍遠不如人意。CO2分子的內在遲緩性是光催化CO2還原的關鍵限制。因此,開發高活性光催化劑已成為研究熱點。二維g-C3N4基光催化劑具有良好的CO2親和力、低CO2活化屏障、較強的氧化還原能力和光穩定性,受到越來越多的關注。
4.3 可見光催化降解
隨著人口的快速增長和工業化的顯著發展,大量有毒、有害和無窮無盡的污染物入侵環境,威脅著人類健康,特別是水中存在的各種難以自然消除或降解的污染物。光催化降解有毒污染物是解決環境問題的綠色經濟途徑,不同種類的g-C3N4被用于提高污染物的光降解效率。g-C3N4的CB 和VB 在光激發的作用下產生電子和空穴,由于空穴具有較強的氧化性,可以直接與有機污染物發生氧化反應。在光催化過程中,有機污染物被降解。Yan 等[48]通過三聚氰胺和氧化硼的熱縮合合成了B 摻雜g-C3N4。獲得的B 摻雜g-C3N4表現出增強的光吸收和染料吸附,促進了光催化RhB 的降解。Fu等[39]通過AgBr/g-C3N4雜化物光還原得到了Ag@AgBr/g-C3N4復合物。g-C3N4的CB 中的剩余電子和AgBr 的VB 中的空穴對甲基橙和RhB 的降解表現出較強的還原和氧化能力。在另一份報告中,Katsumata 等[49]獲得了由Ag3PO4,Ag 和g-C3N4構成的Z 型異質結,用于甲基橙的高效光催化脫色。
5 結 論
總之,由于其可調節的電子能帶結構、優異的穩定性、廉價的材料、易于合成和豐富的自然特性,不含金屬的聚合物g-C3N4可被視為產氫、降解有機/無機污染物和CO2光還原的有效光催化劑。然而,由于其結晶度差、比表面積低、可見光吸收能力有限和電荷載流子復合速率高等問題限制了其光活性。因此,對于g-C3N4的改性,如形貌調控、金屬或非金屬雜原子摻雜和與其他光催化半導體的偶聯等方法,可用于增加其光活性。本文討論了改性的g-C3N4光催化劑在光催化產氫、CO2光還原和降解不同有機污染物方面的應用,并表明改性后g-C3N4光催化劑在電學和光學性能方面得到了顯著的提高,獲得了顯著的量子效率和光催化性能。然而,該領域仍存在若干挑戰需要研究者們繼續重點和深入探究,包括:
(1) 在催化材料合成方面,需要開發更高效的光催化劑合成方法,并探究不同因素對合成方法、光催化性能和形貌的影響。此外,還應該考慮前驅體的選擇、溫度以及pH 等因素,以避免對催化性能產生負面影響;
(2) 改性g-C3N4基光催化劑存在氧化還原能力較低的問題,因此,其活性遠不能滿足實際應用的要求。為了解決這個問題,需要進一步進行研究和改進,以提高其催化效率和活性;
(3) 改性后g-C3N4光催化劑的催化機制驗證存在問題,因為光生電荷的分離和遷移發生的很快,現有的技術無法檢測載流子的具體反應過程,因此開發有效且直觀的監測技術顯得十分重要。
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
