BHA 浮選錫石體系中鋅組分活化機理的量子化學計算研究
2023-09-19 00:41:10代龍富郝佳美秦曉艷高虎林
金屬礦山 2023年8期
代龍富 劉 建,2 李 達 郝佳美 秦曉艷 高虎林
(1.昆明理工大學國土資源工程學院,云南 昆明 650093;2.省部共建復雜有色金屬資源清潔利用國家重點實驗室,云南 昆明 650093)
錫是人類歷史上最早被利用的金屬之一,同時也是我國重要的戰略金屬之一[1-2]。錫具有無毒、熔點低、耐腐蝕、質地軟、延展性好等優良性質,被廣泛應用于電子、化工、冶金、機械、食品包裝、原子能及航天工業等領域[3-4]。錫石中理論錫含量為78.8%,是地殼中最常見的含錫礦物,且被認為是唯一可利用的錫礦物。錫石由四方結構的二氧化錫組成,具有高密度、強硬度和高脆性,使其適用于重選分離[5-7]。但隨著高品質錫石的逐漸枯竭,低品位細粒錫石和重選尾礦正在成為錫石浮選原料的主要來源;由于錫石浮選捕收劑的發展及應用,使得這類錫石的有效回收成為可能[8-11]。量子化學計算是研究礦物的晶體結構、電子性質及表面吸附的有效方法,在礦物表面吸附化學等領域有著非常重要的應用[12-16]。根據密度泛函理論可獲得礦物表面的微觀結構及物理化學信息,如通過前線軌道分析可以判斷得失電子中心、HOMO 與LUMO 分布情況、軌道組成等信息;利用電荷布居分析可以獲得各原子電荷差異,進而判斷物質的反應活性中心;利用反應物與生成物最優結構的能量,計算得到相互作用能,進一步判斷反應物之間作用能力強弱[17-21]。這為研究鋅組分在錫石的BHA 浮選體系中的作用機理提供了有力的幫助。
在錫石浮選中,金屬離子有著非常重要的作用,其中鉛離子被廣泛應用于羥肟酸類浮選錫石,但其具有很強的生物毒性,難以進行生物化學降解,從而導致生物化學和環境問題。因此,有學者已經在尋找用于錫石浮選的替代金屬離子[22-30]。鋅的生物毒性和環境影響遠小于鉛,研究鋅組分是否具有在羥肟酸類捕收劑存在下活化錫石的潛力是十分必要的。CAO[31]研究了鋅組分在BHA 浮選錫石體系中的活化作用,結果表明,鋅組分的存在增加了BHA 在錫石表面的吸附量,且鋅組分的活化效果高于鉛組分,錫石最大的浮選回收率可達90.54%。迄今為止并沒有從原子微觀角度對鋅組分活化錫石表面的作用機理進行系統詳細的探究,BHA 浮選體系中鋅組分活化錫石表面的量子化學研究工作亟須進行。
為了揭示鋅組分在BHA 浮選錫石體系中的活化機理,本研究基于密度泛函理論,建立了錫石表面上鋅組分和BHA 的共吸附模型,并采用第一性原理計算方法研究了鋅組分在BHA 浮選錫石體系中的作用機理,從原子微觀角度定性研究了鋅組分吸附于錫石表面后的電子性質、電荷轉移情況及吸附性能,為鋅組分活化錫石浮選的實踐提供重要的理論指導。
1 模型建立及計算方法
1.1 計算方法
采用Material Studio 中CASTAP 模塊進行所有的結構優化、能量計算和性質分析。根據前人經驗[11,32-34],計算中交換關聯函數采用廣義梯度近似(GGA)下的PBE 梯度修正函數,采用Customized 贗勢描述離子核和價電子之間的相互作用,價電子平面波函數截斷能(Energy cutoff)設置為450 eV,所有計算均在倒易空間中完成,k 點(k-point)網絡設置為2×2×1,自洽場收斂精度設置為2.0×10-6eV/atom;原子間相互作用力(Max.force)收斂標準設置為0.01 eV/?(1 ?=0.1 nm);晶體內應力的收斂標準(Max.stress)設置為0.2 GPa;最大循環(Max.iterations)設置為500。由于錫石是非磁性礦物,后續所有計算過程中均采用非自旋進行計算。
吸附能是一個或多個分子在吸附界面上方運動時,在其速率由大變小并最終吸附在吸附界面上的過程中釋放出的能量。不同分子或離子與相同礦物表面作用時的吸附能有差異,由此可憑借吸附能來判斷藥劑與礦物表面的吸附作用,吸附能為負值時說明吸附可以自發進行,吸附能的值越小代表著吸附越穩定;吸附能為0 或正值時表明吸附不能自發進行。藥劑分子在礦物表面上的吸附能計算如下式所示:
ΔE=Ecom-Esurf-Erea,
式中:ΔE為吸附能,Ecom為吸附絡合物的總能量,Esurf為表面結構的總能量,Erea為吸附質的總能量,單位均為kJ/mol。
1.2 錫石晶體及表面的構建及優化
錫石化學式為SnO2,屬于四方晶系的氧化礦物,具有金紅石構型,通常為雙錐狀或細長柱狀的短柱體[5-6]。通過幾何優化得到的錫石晶體單胞的晶格參數如表1 所示,與實驗值相對誤差均小于0.01%,可進行后續計算。
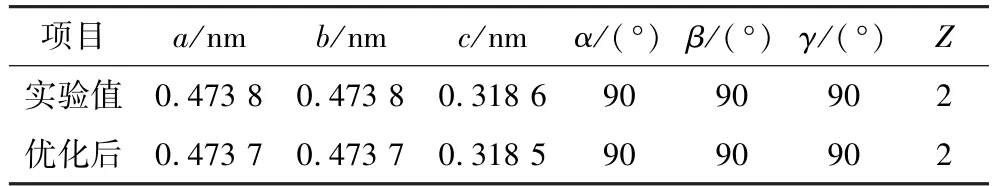
表1 經幾何優化后錫石晶體單胞模型的晶格參數Table 1 Lattice parameters of cassiterite crystal unit cell model after geometric optimization
錫石作為典型的AX2型氧化礦物,晶體結構是六方最緊密堆積,其表面疏水性較弱。通常,在外力的作用下,礦物一般會沿著平行于層間距較大的面、陰陽離子電性中和的面、兩層離子相鄰的面和化學鍵合強度最弱的方向產生破碎。研究表明(110)表面的表面能與斷鍵密度遠小于(101)面和(111)面,是錫石表面暴露最多的表面,可將(110)表面作為錫石顆粒的代表性表面[32]。因此,本研究均在錫石的(110)表面上進行。考慮到后續鋅組分與BHA 分子在錫石表面的吸附,采用2×2×2 的超胞模型,設置真空層為1.5 nm,共3 個原子層,共包含24 個Sn 原子和48 個O 原子共72 個原子;為提高計算效率,固定底部兩層原子。
圖1 為經幾何優化后的錫石(110)面模型,錫石(110)面呈現出1 個雙配位的O(頂位)原子與2 個六配位的Sn 原子結合在1 個平面上,和1 個三配位的O(底位)原子與2 個五配位的Sn 原子和1 個六配位的Sn 原子相連接,整體呈一個類似凸凹的結構。
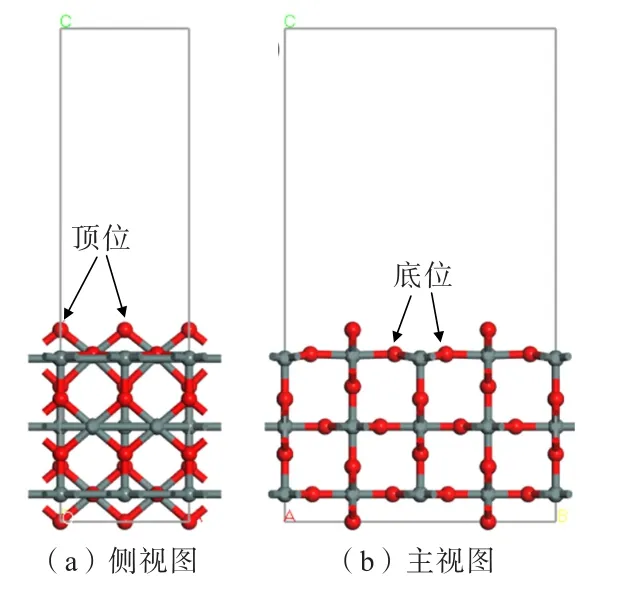
圖1 經幾何優化后的錫石(110)面模型Fig.1 Geometrically optimized cassiterite (110)surface model
1.3 藥劑分子模型的構建及優化
CAO 等[31]研究結果表明:以BHA 和ZnSO4浮選錫石時,最佳pH 條件為7.5 ~8.0。由溶液化學計算結果可知,溶液中的鋅組分在pH 為7.5~8 時主要以游離的Zn(OH)+的形式存在[31,35]。溶液中的BHA在pH=7.5~8 時主要以游離的BHA 分子的形式存在(平衡濃度:pKa=8.5 ~9.0)[31,36]。因此,本文以Zn(OH)+代表鋅組分與BHA 分子進行計算。在3D模型中構建Zn(OH)+分子與BHA 分子結構模型,再采用CASTAP 模塊對Zn(OH)+分子與BHA 分子的結構模型分別進行結構優化,計算中交換關聯函數采用廣義梯度近似(GGA)下的PBE 梯度修正函數,采用Fine 贗勢描述離子核和價電子之間的相互作用。優化后的Zn(OH)+分子與BHA 分子的結構如圖2所示。
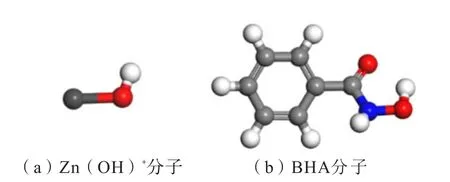
圖2 經幾何優化后的藥劑分子結構模型Fig.2 Molecular structure model of reagent after geometric optimization
2 試驗結果與討論
2.1 錫石(110)面的態密度分析
首先計算了未吸附藥劑分子前的錫石(110)面各原子分態密度及總體態密度,結果如圖3 所示。
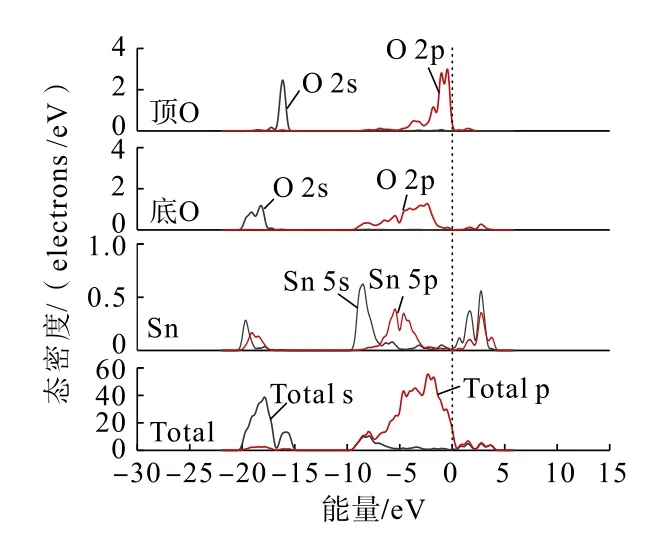
圖3 錫石(110)面各原子分態密度及總體態密度Fig.3 Partial and total density of states of each atom on the surface of cassiterite (110)
通過錫石(110)解離面的態密度分析可以發現:在-20~-15 eV 價帶主要由O(頂位)原子與O(底位)原子的2s 軌道以及少量Sn 的5s、5p 軌道組成;在-9~1 eV 價帶主要由O(頂位)原子與O(底位)原子的2p 軌道以及部分Sn 的5s、5p 軌道組成;在1 ~4 eV價帶主要由Sn 的5s、5p 軌道。在費米能級附近主要由O(頂位)原子的2p 軌道組成,說明O(頂位)原子的活性較強,容易參與反應。
2.2 鋅組分在錫石表面上的吸附情況
在礦物浮選中,藥劑分子在礦物表面上的吸附取決于吸附位點及其活性與吸附形式,2.1 節已說明錫石(110)表面的O(頂位)原子活性較強。故本節根據Zn(OH)+在錫石(110)面O(頂位)原子位點上不同的吸附形式,分別計算了Zn(OH)+在錫石(110)面O(頂位)原子位點上單橋形式與雙橋形式的吸附情況。
2.2.1 吸附能
圖4 為Zn(OH)+在錫石(110)面O 原子位點上不同形式的吸附模型。單橋形式吸附時,Zn(OH)+在錫石表面1 個O(頂位)原子斜上方,與1 個O(頂位)原子形成線吸附;雙橋形式吸附時,Zn(OH)+吸附在錫石(110)面的2 個O(頂位)原子上,形成面吸附。計算得到的吸附能如表2 所示。
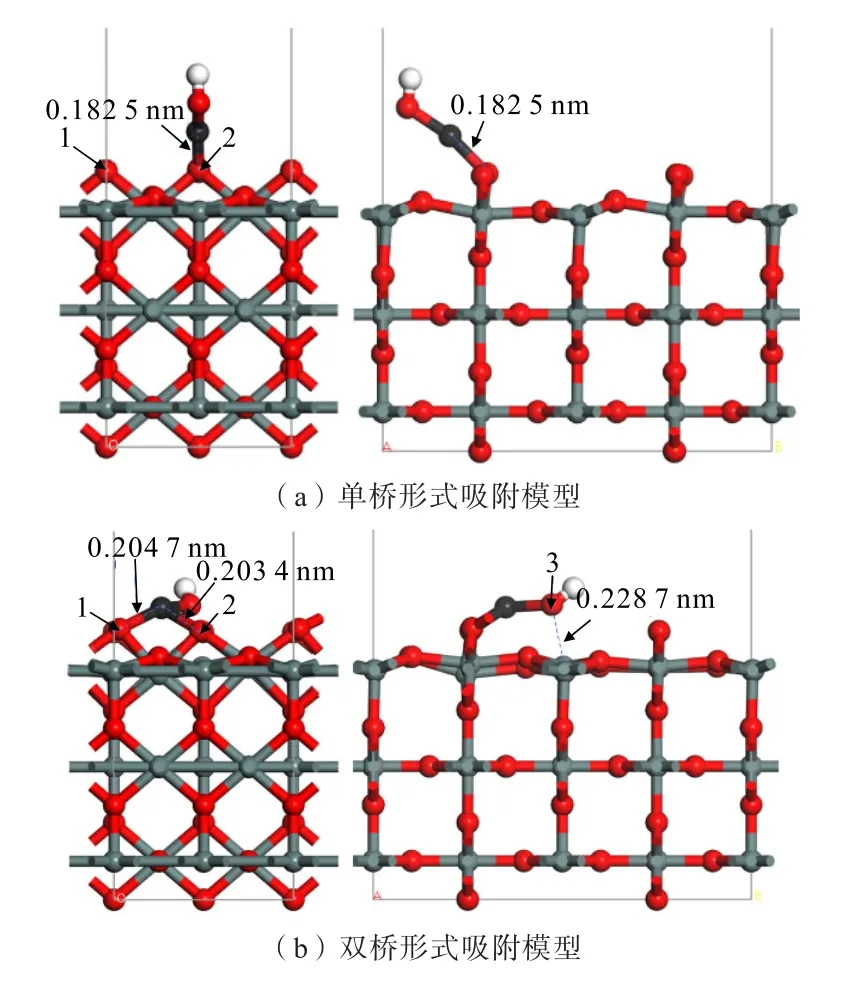
圖4 Zn(OH)+在錫石(110)面上不同形式的吸附模型Fig.4 Different adsorption models of Zn(OH)+on cassiterite (110) surface

表2 Zn(OH)+在錫石(110)面上不同吸附形式的吸附能及鍵長Table 2 Different adsorption forms of adsorption energy and bond length of Zn(OH)+on cassiterite (110) surface
從表2 可知,Zn(OH)+在錫石(110)表面上單橋與雙橋形式吸附時的吸附能分別為-532.39、-580.64 kJ/mol,吸附能均為負值,說明Zn(OH)+可在錫石(110)面上自發吸附。
由圖4 可知,單橋形式吸附的最佳吸附構型中,Zn(OH)+中Zn 原子與錫石表面1 個O(頂位)原子形成的Zn—O 鍵長為0.182 5 nm,小于Zn、O 原子的半徑之和(0.205 nm),說明了Zn(OH)+在錫石表面上的O(頂位)原子發生了化學吸附。雙橋形式吸附的最佳吸附構型中,Zn—O 鍵長分別為0.204 7、0.203 4 nm;Zn(OH)+中Zn 原子與兩個O(頂位)原子生成的Zn—O 鍵長小于Zn、O 原子的半徑之和(0.205 nm),發生了化學吸附;此外,Zn(OH)+中的O原子向錫石(110)面上的Sn 原子處發生偏移,原子間距為0.228 7 nm,大于Sn、O 原子的半徑之和(0.224 0 nm),說明了Zn(OH)+中的O 原子與錫石(110)面上的Sn 原子未發生化學吸附,未形成穩定的Sn—O 鍵。
總體比較Zn(OH)+分別與錫石(110)面上不同形式的吸附情況,發現Zn(OH)+均能以不同形式與錫石(110)面發生吸附。其中,雙橋形式的吸附能更負,吸附作用更穩定,且Zn(OH)+中O 原子向錫石(110)面上的Sn 原子處發生偏移,形成了更穩定的面吸附形式。說明Zn(OH)+在錫石(110)面上的吸附主要是發生面吸附的雙橋形式。
2.2.2 態密度
為了從原子角度直觀分析Zn(OH)+在錫石(110)面上不同形式的吸附情況,利用態密度分析Zn(OH)+在錫石(110)面O 原子(圖中O 原子序號見圖4)位點上以不同形式吸附前后,各軌道組成的貢獻情況。吸附前后的錫石(110)面上O 原子與Zn原子的態密度分別如圖5、圖6 和圖7 所示。
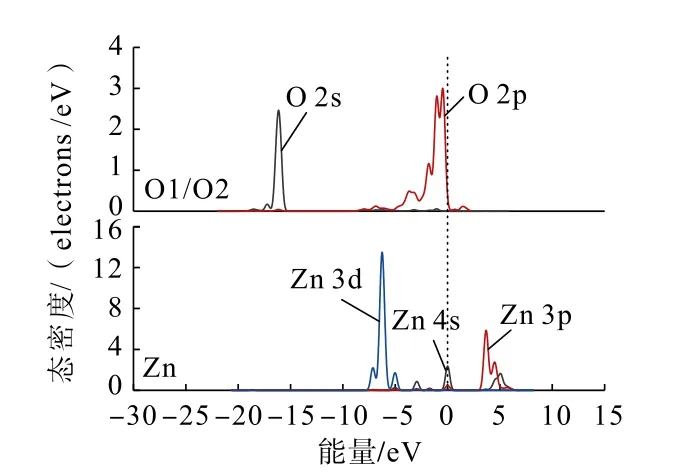
圖5 吸附前錫石(110)面上O 原子與Zn(OH)+中Zn 原子的分態密度Fig.5 Partial density of states between O atoms on cassiterite (110) surface and Zn atoms in Zn(OH)+before adsorption
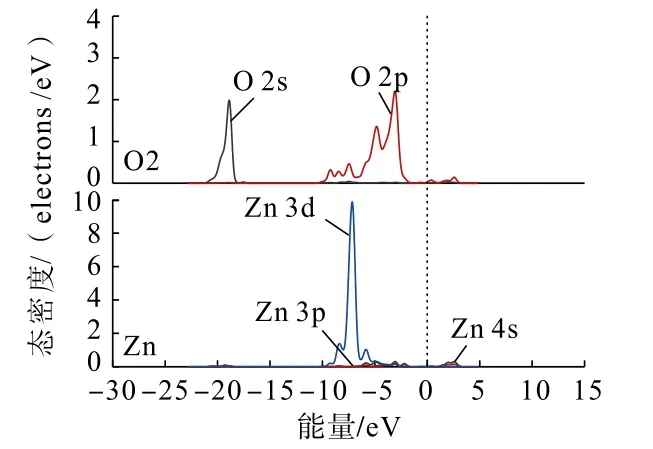
圖6 單橋形式吸附后錫石(110)面上O 原子與Zn(OH)+中Zn 原子的分態密度Fig.6 Partial density of states between O atoms on cassiterite (110) surface and Zn atoms in Zn(OH)+after single bridge adsorption
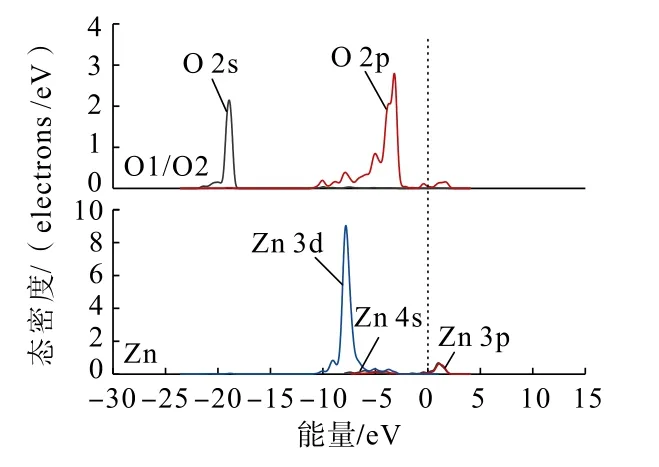
圖7 雙橋形式吸附后錫石(110)面上O 原子與Zn(OH)+中Zn 原子的分態密度Fig.7 Partial density of states between O atoms on cassiterite (110) surface and Zn atoms in Zn(OH)+after double bridge adsorption
通過對比Zn(OH)+在錫石(110)面O(頂位)原子位點上以不同形式吸附前后的態密度可以發現:Zn(OH)+以不同形式吸附后,錫石表面O 原子的2s、2p 軌道與Zn 原子的3 d 軌道均向低能量方向分別移動,說明在加入Zn(OH)+后Zn、O 原子經成鍵相互作用生成Zn—O 鍵,使得Zn(OH)+能夠吸附在錫石(110)面。雙橋形式吸附時,Zn 原子的3d 軌道較雙橋形式吸附時向低能量方向移動更深,說明了Zn(OH)+以雙橋形式吸附在錫石(110)面較為穩定。
Zn(OH)+以雙橋形式吸附在錫石(110)面時,Zn(OH)+中O 會向錫石(110)面上的Sn 原子發生偏移。Zn(OH)+在錫石(110)面O 原子位點上發生雙橋形式的吸附前后,對Zn(OH)+中O 原子與錫石表面Sn 原子進行態密度分析,態密度分別如圖8 與圖9 所示。
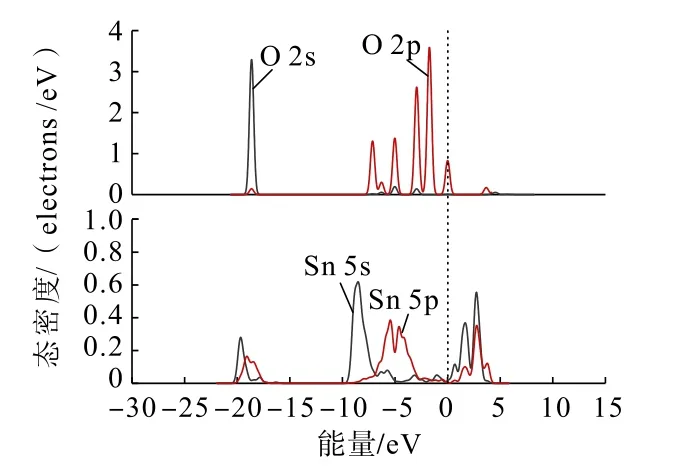
圖8 Zn(OH)+中O 原子與錫石(110)面Sn 原子的分態密度Fig.8 Partial density of states between O atoms in Zn(OH)+and Sn atoms on cassiterite (110) surface
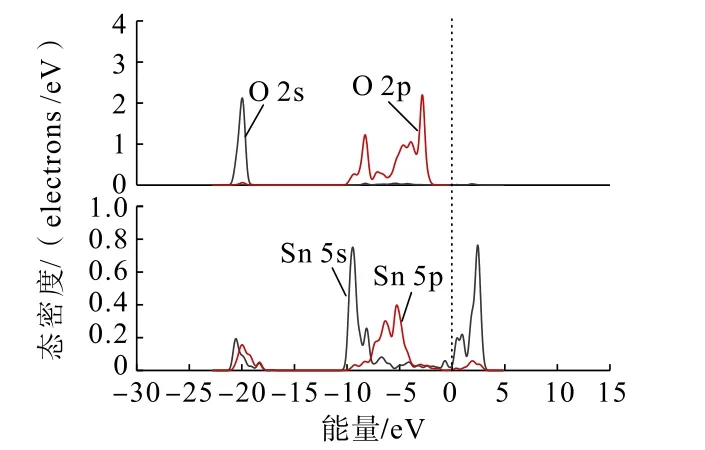
圖9 雙橋形式吸附后Zn(OH)+中O 原子與錫石(110)面Sn 原子的分態密度Fig.9 Partial density of states between O atoms in Zn(OH)+and Sn atoms on cassiterite (110) surface after double bridge adsorption
通過對比Zn(OH)+在錫石(110)面O 原子位點上以雙橋形式吸附前后的態密度可以發現:Zn(OH)+吸附后,Zn(OH)+中O 原子的2s、2p 軌道與錫石表面Sn 原子的5s、5p 軌道均向低能量方向移動不明顯,在費米能級附近的O 原子的2p 軌道峰數減少,越過費米能級在-4 ~-10 eV 處與Sn 原子的5s、5p 軌道未發生交疊,說明了在加入鋅組分后,Zn(OH)+中O 原子未吸附在錫石表面的Sn 原子上,未形成Sn—O 鍵。這與Zn(OH)+在錫石(110)面上的吸附結果一致。
2.2.3 Mulliken 布居分析
在分子作用體系中,為了研究各原子的電荷分布、轉移以及成鍵特性等性質,常通過Mulliken 布居進行分析判斷。通過對Zn(OH)+以不同形式吸附在錫石(110)面上O 原子前后模型進行計算,得到了Mulliken 鍵布居與鍵長,如表3 所示(O 原子對應序號位置見圖4)。

表3 Zn(OH)+以不同吸附形式吸附在錫石(110)面的Mulliken 鍵布居與鍵長Ta ble 3 Mulliken bond population and bond length of Zn(OH)+ adsorbed on cassiterite(110) surface in different adsorption forms
從表3 可以看出,Zn(OH)+以單橋形式吸附后,錫石(110)表面形成的Zn—O2 化學鍵的Mulliken 鍵布居和鍵長分別為0.32、0.182 5 nm,顯然在Zn(OH)+以單橋形式吸附錫石(110)表面的過程中形成了穩定的共價鍵,可以認為Zn(OH)+以單橋形式與錫石表面的O2原子發生吸附,形成直線吸附結構。Zn(OH)+以雙橋形式吸附后,錫石(110)表面形成的Zn—O1、Zn—O2 化學鍵的Mulliken 鍵布居和鍵長分別為0.35、0.204 7 nm 與0.32、0.203 4 nm,其中Zn—O1 與Zn—O2 兩者的Mulliken 鍵布居和鍵長較為接近,成鍵穩定性好,各化學鍵的共價性差異較小,可以認為Zn(OH)+以雙橋形式吸附在錫石(110)面時,受空間因素影響,與O1、O2 原子同時吸附,形成更為穩定的面螯合結構。
表4 為Zn(OH)+以不同吸附形式吸附在錫石(110)面各原子的Mulliken 電荷布居(O 原子對應序號位置見圖4)。
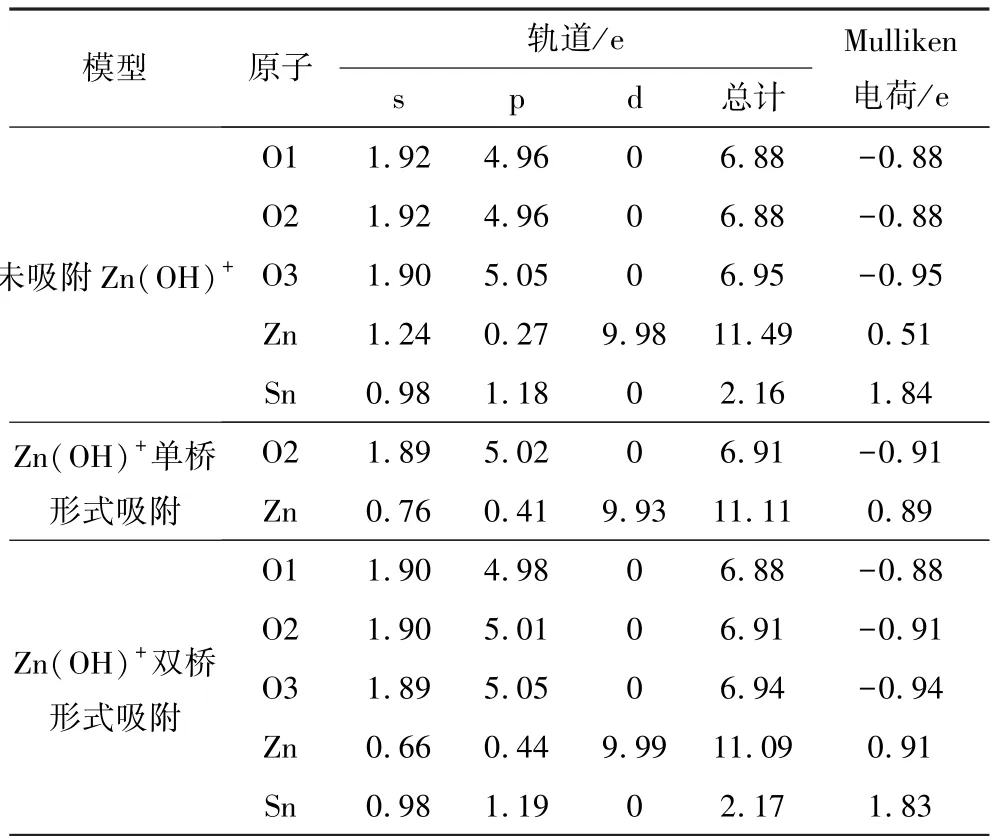
表4 Zn(OH)+以不同吸附形式吸附在錫石(110)面各原子的Mulliken 電荷布居Table 4 Mulliken charge population of Zn(OH)+adsorbed on each atom of cassiterite (110) surface in different adsorption forms
由表4 可知,Zn(OH)+以不同形式吸附在錫石(110)面O 原子位點上后,Zn 原子電荷總量均降低,其中4s 和3p 軌道的電荷發生了較大的變化,3d 軌道的電荷幾乎無任何變化,結合Zn(OH)+吸附前后Zn 原子的態密度結果進行分析,在吸附過程中,Zn原子發生電荷轉移可能是因為3d 電子轉移后,4s 或3p 再次向3d 轉移。同時,O 原子電荷總量升高,其2s、2p 軌道在吸附前后均發生了變化。在雙橋形式吸附時,Sn 原子電荷總量與Sn 原子發生直接作用的O3 原子電荷總量均變化不明顯,說明在吸附過程中,Sn 原子與Zn(OH)+中O3 原子未發生化學吸附。比較兩種不同吸附形式的吸附模型可知,雙橋形式吸附后各原子的轉移電荷總數要大于單橋形式吸附后各原子的轉移電荷總數,說明了Zn(OH)+吸附錫石時雙橋形式吸附更穩定,這與吸附能和態密度的討論結果相一致。
2.2.4 差分電荷密度分析
通過態密度及Mulliken 布居分析發現:鋅組分在錫石(110)表面O 原子位點上吸附時,O、Zn 原子之間發生了電荷轉移并形成了新的Zn—O 鍵,為了直觀地分析電子的分布密度并判斷原子之間的成鍵情況,對Zn(OH)+以不同形式吸附錫石(110)面后模型進行了差分電荷密度分析,結果如圖10 所示。當兩原子之間最低電荷密度顏色比背景板的顏色更深時,說明兩原子之間成共價鍵,反之,成離子鍵;同時也可通過顏色的空間分布判斷原子之間得失電子的情況。
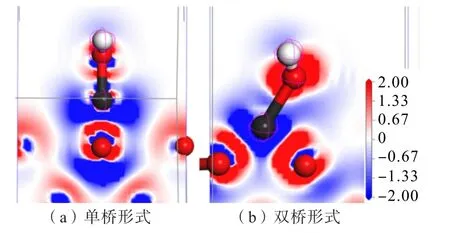
圖10 Zn(OH)+以不同形式吸附錫石(110)面后的差分電荷密度Fig.10 Differential charge density diagram of Zn(OH)+adsorbed cassiterite (110) surface in different forms
通過差分密度圖可知:Zn(OH)+在錫石(110)面的O 位點以不同形式吸附后,Zn 原子與錫石(110)面的O(頂位)原子之間均出現了電荷交互,說明Zn、O(頂位)原子之間發生了電子的轉移,生成了Zn—O共價鍵;又由于Zn(OH)+之間原本就存在的化學鍵,因此也出現了電荷交互的作用。對比圖10(a)與圖(b)可以發現,Zn(OH)+在錫石(110)面的O 位點以雙橋形式吸附時,Zn、O 原子之間最低電荷密度顏色比單橋形式的顏色更深,轉移電荷量更多,說明Zn(OH)+吸附錫石時以雙橋形式吸附更穩定,與前文討論的結果相一致。
2.3 BHA 在鋅組分作用后的錫石表面上吸附
由2.2 可知,Zn(OH)+以雙橋形式吸附在錫石(110)面上吸附能較低,吸附穩定性好,故本節分別計算了BHA 與錫石(110)面、BHA 與Zn(OH)+以雙橋形式作用后的錫石(110)面的吸附情況,討論Zn(OH)+的加入對BHA 浮選錫石體系的作用機制。優化后的吸附模型如圖11 所示,吸附能計算結果如表5 所示。
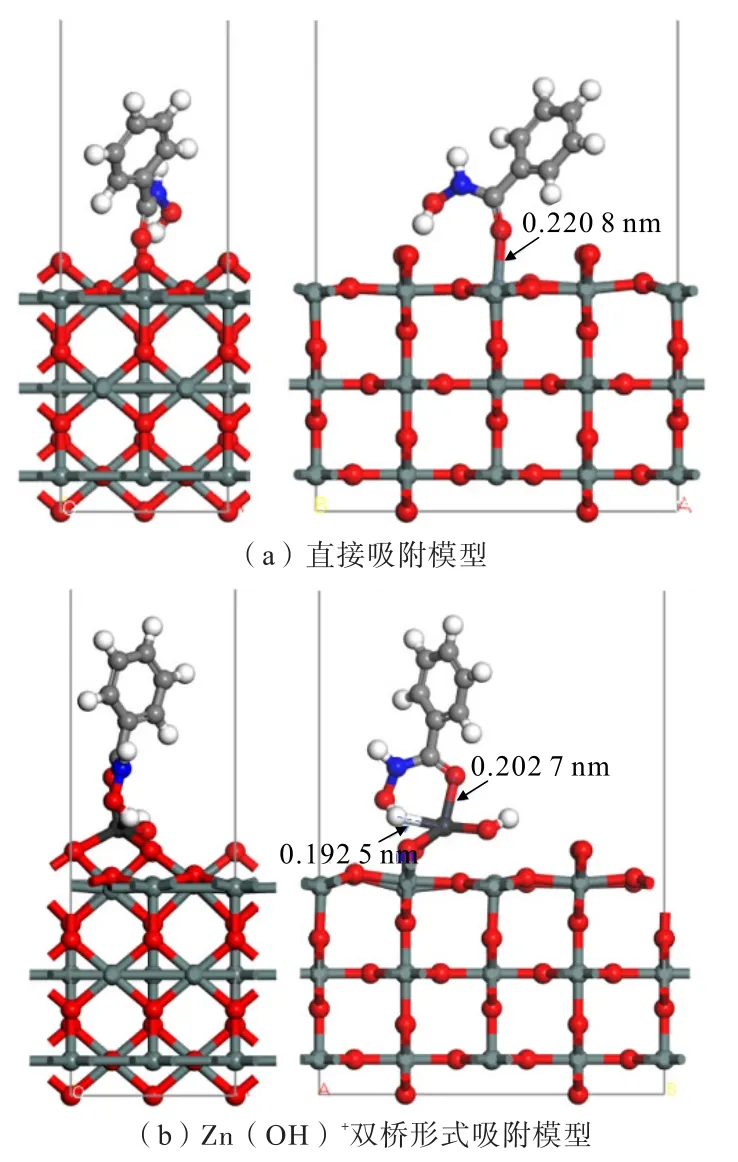
圖11 Zn(OH)+加入前后,BHA 吸附于錫石(110)面的吸附情況Fig.11 The adsorption of BHA on cassiterite (110) surface before or after the addition of Zn(OH)+

表5 Zn(OH)+加入前后,BHA 吸附于錫石(110)面的吸附能Table 5 Adsorption energy of BHA adsorbed on cassiterite (110) surface before or after the addition of Zn(OH)+
表5 表明,BHA 與錫石(110)面直接吸附時,吸附能為-221.23 kJ/mol,吸附能為負,說明BHA 可自發地在錫石表面吸附;而加入Zn(OH)+對錫石(110)面作用后,BHA 與作用后的錫石(110)面吸附能為780.97 kJ/mol,為負值,且較BHA 與錫石(110)面直接吸附的吸附能更負,說明Zn(OH)+的存在有利于BHA 在錫石表面的吸附。如圖11 所示的吸附模型,BHA 與錫石(110)面直接吸附時,BHA 分子中雙鍵O原子與錫石(110)面的Sn 原子直接形成單鍵吸附;而加入Zn(OH)+對錫石(110)面作用后,BHA 分子中雙鍵O 原子和—OH 基團與錫石(110)面上吸附的Zn(OH)+中的Zn 原子的作用距離分別為0.202 7、0.192 5 nm,形成了穩定的五元環螯合結構。總的來看,Zn(OH)+預先作用在錫石(110)面后,有利于BHA 在錫石(110)面形成穩定的五元環螯合結構進行吸附。
3 結 論
(1)錫石(110)面的態密度分析結果顯示,在費米能級附近主要由O(頂位)原子的2p 軌道組成,說明O(頂位)原子的活性較強,容易參與反應。
(2) 以Zn(OH)+代表的鋅組分均可在錫石(110)面以不同形式與O 原子位點間發生吸附作用,Zn 原子與O 原子間均發生電荷轉移,軌道向低能量方向移動,形成Zn—O 鍵;在雙橋形式吸附時,Zn 原子的3d 軌道移動更負,轉移的電荷量更多,形成的Zn—O 鍵穩定性更好。說明了Zn(OH)+在錫石(110)面上主要是以面螯合吸附的雙橋形式吸附。
(3)BHA 可自發通過羥肟酸基團中的雙鍵O 原子與錫石(110)面的Sn 原子吸附連接成鍵,然而受空間位阻影響,BHA 分子吸附在錫石表面上未形成穩定的螯合結構。加入鋅組分對錫石(110)面作用后,BHA 中雙鍵O 原子和—OH 基團與錫石(110)面上吸附的Zn(OH)+中的Zn 原子形成穩定的五元環螯合吸附,提高了BHA 在錫石(110)面的吸附能力和穩定性。
