去氫木香內(nèi)酯在內(nèi)生真菌Y0 液體發(fā)酵中的生物轉(zhuǎn)化研究
2023-07-12 01:44:14鄒秋萍劉明秀李寶晶何紅平汪偉光李艷平
云南中醫(yī)學(xué)院學(xué)報 2023年3期
關(guān)鍵詞:結(jié)腸癌
郭 晶,陳 帥,鄒秋萍,劉明秀,李寶晶,何紅平,汪偉光,李艷平*
(1. 云南中醫(yī)藥大學(xué)中藥學(xué)院暨云南省南藥可持續(xù)利用重點實驗室,云南 昆明 650500;2. 云南中醫(yī)藥大學(xué)云南省傣醫(yī)藥與彝醫(yī)藥重點實驗室,云南 昆明 650500;3. 云南民族大學(xué)國家民委民族藥內(nèi)生菌天然產(chǎn)物合成生物學(xué)重點實驗室,云南 昆明 650500)
微生物轉(zhuǎn)化(microbial transformation,MT)是通過微生物細胞將復(fù)雜的底物進行結(jié)構(gòu)修飾,也就是利用微生物代謝過程中產(chǎn)生的某個或某一系列的酶對底物特定部位進行的催化反應(yīng)[1]。近年來,微生物轉(zhuǎn)化技術(shù)因具有反應(yīng)類型廣、特異性強、副產(chǎn)物少、反應(yīng)條件溫和可控、環(huán)保無污染等優(yōu)點,已逐漸成為開發(fā)高活性、低毒性新藥的一條高效途徑[2-3]。利用微生物獨特而多樣的酶系統(tǒng),以中藥活性成分作為底物,將微生物轉(zhuǎn)化技術(shù)應(yīng)用于中藥活性成分的結(jié)構(gòu)修飾,可獲得更多結(jié)構(gòu)新穎活性顯著的天然產(chǎn)物,可為現(xiàn)代中藥研發(fā)提供一種新的有效路徑。Ma 等[4]報道了Mucor polymorphosporus AS 3.3443 能夠通過有效催化脫氫,使廣木香內(nèi)酯的C11-C13雙鍵專一性還原轉(zhuǎn)化為11α,13-二氫脫氫廣木香內(nèi)酯(11α,13-dihydrodehydrocostuslactone)。劉偉[5]發(fā)現(xiàn)Cunninghamella elegans AS 3.2028 能夠轉(zhuǎn)化莪術(shù)二酮為環(huán)氧化產(chǎn)物8,9-環(huán)氧化莪術(shù)二酮。Arakawa 等[6]報道了土曲霉和黑曲霉(Aspergillus terreus)(A. niger)對芽孢素A 的生物轉(zhuǎn)化,獲得了三種具有新穎結(jié)構(gòu)骨架的倍半萜衍生物。與原始天然產(chǎn)物相比,真菌轉(zhuǎn)化產(chǎn)物對HL-60細胞的細胞毒性更低。
木香(Aucklandia lappa)具有解痙、降壓及抗菌作用[7],是多種中成藥的組方之一,例如香砂養(yǎng)胃片,其中主要活性成分是木香烴內(nèi)酯和去氫木香內(nèi)酯,其中木香烴內(nèi)酯0.400~1.463 mg/片,去氫木香烴內(nèi)酯0.512~0.983 mg/片[8]。木香烴內(nèi)酯為倍半萜內(nèi)酯類化合物,結(jié)構(gòu)中α-亞甲基-γ-內(nèi)酯是其具有抗腫瘤活性的必要基團,天然存在的倍半萜內(nèi)酯類化合物雖然種類多,但含量較少,相關(guān)研究也較少。去氫木香內(nèi)酯具有愈創(chuàng)木烷骨架[9],被認為是生物合成倍半萜內(nèi)酯的重要中間體,但因其較低的水溶性限制了它在臨床上的應(yīng)用,而通過具有立體和區(qū)域?qū)R恍缘纳镛D(zhuǎn)化作用能夠有效改善其溶解性和生物活性[10]。Choi 等[11]研究了去氫木香內(nèi)酯的抗腫瘤活性,考察了其對人類乳腺癌細胞和卵巢癌細胞的作用,發(fā)現(xiàn)去氫木香內(nèi)酯具有潛在的抗腫瘤活性。Park 等[12]的研究也表明去氫木香內(nèi)酯和木香烴內(nèi)酯還具有顯著抑制人類乳腺癌細胞MCF-7 和MDA-MB-453 的活性。Liu 等[13-14]主要研究了木香烴內(nèi)酯對人類肝癌細胞(Hepatic Cell Carcinoma)有絲分裂時細胞活力的影響,表明木香烴內(nèi)酯可以降低人類肝癌細胞(HCC)有絲分裂時細胞活力,抑制細胞周期。以上研究表明木香中以木香烴內(nèi)酯和去氫木香內(nèi)酯為主的衍生物具有較好的生物活性和開發(fā)利用前景,而通過生物轉(zhuǎn)化的方式利用富含豐富酶系的真菌對木香烴內(nèi)酯和去氫木香內(nèi)酯進行轉(zhuǎn)化,有可能獲得毒性較低,生物活性好的結(jié)構(gòu)修飾產(chǎn)物[15]。本研究將探索木香烴內(nèi)酯和去氫木香內(nèi)酯在真菌Y0 中生物轉(zhuǎn)化的情況。同時通過MTT 法檢測轉(zhuǎn)化產(chǎn)物對結(jié)腸癌細胞SW620 的細胞毒性,并根據(jù)網(wǎng)絡(luò)藥理學(xué)預(yù)測轉(zhuǎn)化產(chǎn)物的作用靶點。
1 材料與方法
1.1 儀器和試劑 立式壓力蒸汽滅菌器(上海博迅實業(yè)有限公司醫(yī)療設(shè)備廠);恒溫振蕩培養(yǎng)箱(常州國華電器有限公司);霉菌培養(yǎng)箱(上海一恒科學(xué)儀器有限公司);超凈工作臺(蘇州安泰空氣技術(shù)有限公司);分析天平;高效液相色譜儀(安捷倫公司);AV-600 Hz 核磁共振儀(德國Bruker 公司)。
乙酸乙酯(分析純)、甲醇(色譜純)、葡萄糖(分析純);木香烴內(nèi)酯、去氫木香內(nèi)酯;PDA(Potato Dextrose Agar 馬鈴薯葡萄糖瓊脂)培養(yǎng)基(上海博微生物科技有限公司);PDB 培養(yǎng)基(新鮮去皮馬鈴薯:葡萄糖:蒸餾水,比例為10 ∶1 ∶50);正相硅膠柱色譜(100~200 目)。
1.2 菌株培養(yǎng)鑒定和生物轉(zhuǎn)化 去氫木香內(nèi)酯和木香烴內(nèi)酯分別在內(nèi)生真菌Y0(Coniothyrium pyrinum)發(fā)酵液進行生物轉(zhuǎn)化研究。用PDA 培養(yǎng)基活化菌株Y0,倒置于28 ℃恒溫培養(yǎng)箱中培養(yǎng),無菌條件下,挑取長勢良好的新鮮菌落放入裝有PDB 培養(yǎng)基離心管,恒溫振蕩培養(yǎng)箱28 ℃、120 rpm 培養(yǎng)。
分別稱取50 mg 去氫木香內(nèi)酯和木香烴內(nèi)酯置于離心管中,加DMSO 溶解,用PTFE 膜過濾,制成25 mg/mL 的化合物溶液。取80 μL 化合物溶液至培養(yǎng)液(50 mL)中,混勻并做好標記,設(shè)置僅含培養(yǎng)基不含化合物溶液的菌液做為空白對照,恒溫振蕩培養(yǎng)箱28 ℃、120 rpm 培養(yǎng)。第1、3、5、7 天分別取發(fā)酵液(2 mL)用等體積乙酸乙酯超聲提取,離心,取上清液旋干,用色譜甲醇溶解后用高效液相檢測,在第3 天有轉(zhuǎn)化產(chǎn)物生成。
1.3 提取、分離純化及鑒定 培養(yǎng)液加入等體積乙酸乙酯超聲提取30 min,離心,取上清液蒸干,濃縮。將濃縮液用硅膠柱色譜分離,用石油醚-乙酸乙酯(15 ∶1~1 ∶1 梯度洗脫),得到Fr 1~Fr 4,F(xiàn)r 2 用高效液相色譜檢測。采用色譜柱Zorbax SB-C18(250 mm×9.4 mm,5 μm);檢測波長:210 nm;柱溫:30 ℃;以乙腈為流動相A,水為流動相B,乙腈/水=40 ∶60(v/v);流速:流速3 mL/min 等度洗脫,得到化合物1(5 mg,tR=16.16 min)和化合物2(0.7 mg,tR=15.57 min)。通過薄層色譜GF254 硅膠板,以10%濃硫酸-乙醇顯色檢識轉(zhuǎn)化產(chǎn)物,根據(jù)化合物波譜數(shù)據(jù)對化合物結(jié)構(gòu)進行解析(1H NMR 和13C NMR)。
1.4 細胞毒檢測方法 收集對數(shù)生長期的結(jié)腸癌SW620 細胞,以5×104個/mL 的密度接種于96 孔板(100 μL/孔),培養(yǎng)24 h 后去除舊培養(yǎng)基,加入含化合物1、化合物2,且濃度為10~50 μg/mL 的新培養(yǎng)基,并孵育24 h,丟棄上清,每孔加入10 μL 5mg/mL MTT 溶液,4 h 后,去除MTT 溶液,加入200 μL 二甲基亞砜(DMSO)。在波長為490 nm 的酶標儀下測定OD 值。根據(jù)以下公式計算細胞存活率:細胞存活率=(A 化合物組-A 調(diào)零組)/(A 對照組-A 調(diào)零組)×100%。
1.5 化合物1 和2 與結(jié)腸癌SW620 細胞靶點預(yù)測與分子對接 使用GeneCards(www.genecards.org)數(shù)據(jù)庫和OMIM(omim.org)數(shù)據(jù)庫獲取結(jié)腸癌SW620細胞相關(guān)靶點;使用Swiss Target Prediction(www.swisstargetprediction.ch)數(shù)據(jù)庫對化合物1 和化合物2 的靶點進行預(yù)測;使用Origin 2021 軟件對兩個化合物的靶點與疾病的靶點做交集維恩圖;使用UniProt(www.uniprot.org)數(shù)據(jù)庫和PDB(www1.rcsb.org)數(shù)據(jù)庫獲取兩個化合物與疾病各自的交集靶點;使用PyMOL2.3.0 軟件和ChemDraw 2020 軟件對靶點蛋白和兩個化合物分別進行分子對接前處理;使用Auto Dock Tools1.5.6 和Auto Dock Vina1.2.0 軟件對兩個化合物和各自的交集靶點蛋白進行分子對接;使用PyMOL 軟件對對接結(jié)果進行可視化處理。
2 結(jié)果
2.1 高效液相色譜檢測圖 去氫木香內(nèi)酯、木香烴內(nèi)酯、Y0 菌液以及去氫木香內(nèi)酯+Y0 菌液和木香烴內(nèi)酯+Y0 菌液制成檢測液,用乙腈-水(10%~100%)梯度分析30 min,檢測結(jié)果如圖1、圖2 所示。

圖1 去氫木香內(nèi)酯、Y0 菌液、去氫木香內(nèi)酯+Y0 菌液高效液相檢測圖
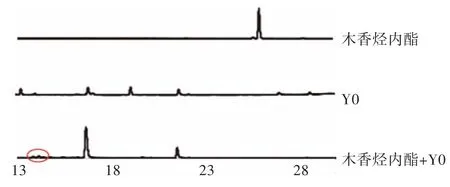
圖2 木香烴內(nèi)酯、Y0 菌液、木香烴內(nèi)酯+Y0 菌液高效液相檢測圖
2.2 去氫木香內(nèi)酯在Y0 液體發(fā)酵液中轉(zhuǎn)化過程圖

圖3 去氫木香內(nèi)酯在Y0 液體發(fā)酵液中轉(zhuǎn)化過程圖
2.3 結(jié)構(gòu)鑒定
2.3.1 化合物1 黃色油狀,C15H20O3,1H-NMR(600 MHz,acetone-d6)δH:2.81(1H,m,H-1),2.11(1H,m,H-2a),2.09(1H,m,H-2b),2.50(1H,m,H-5),3.83(1H,t,J = 9.5 Hz,H-6),1.78 (1H,dd,J = 10.4,9.5 Hz,H-7),2.03(2H,m,H-8),2.50(1H,m,H-9a),1.78(1H,m,H-9b),2.27(1H,m,H-11),1.13(3H,d,J=7.0 Hz,H-13),5.22(1H,m,H-14a),5.21(1H,m,H-14b),4.87(1H,m,H-15a),4.71(1H,m,H-15b).13CNMR(150 MHz,acetone-d6)δC:44.3(C-1),40.7(C-2),74.5(C-3),156.7(C-4),50.5(C-5),85.8(C-6),50.6(C-7),33.2(C-8),39.0(C-9),151.3(C-10),42.2(C-11),178.5(C-12),13.5(C-13),111.0(C-14),112.0(C-15). 以上波譜數(shù)據(jù)與文獻報道基本一致[16],故鑒定化合物1 為(1S,3S,5S,6S,7S,11S)-3- hydroxyl-11,13-dihydrodehydrocostuslactone。
2.3.2 化合物2 無色針晶,C15H20O3,1H-NMR(600 MHz,acetone-d6)δH:2.81(1H,m,H-1),1.92(2H,m,H-2),4.60(1H,m,H-3),2.81(1H,m,H-5),3.96(1H,m,H-6),2.11(1H,m,H-7),1.36(1H,dd,J =12.6,4.6 Hz,H-8a),1.79 (1H,m,H-8b),2.00(1H,dd,J=12.6,4.6 Hz,H-9a),1.92(1H,m,H-9b),2.63(1H,m,H-11),1.10(3H,d,J = 7.8 Hz,H-13),5.24(1H,m,H-14a),5.22(1H,m,H-14b),4.87(1H,s,H-15a),4.73(1H,s,H-15b).13C-NMR(150 MHz,acetone-d6)δC:44.2(C-1),39.8(C-2),74.4(C-3),156.7(C-4),45.4(C-5),85.8(C-6),51.0(C-7),29.6(C-8),38.8(C-9),151.3(C-10),41.0(C-11),179.7(C-12),11.6(C-13),111.1(C-14),111.9(C-15). 以上波譜數(shù)據(jù)與文獻報道基本一致[16],故鑒定化合物2 為(1S,3S,5S,6S,7S,11R)- 3-hydroxyl-11,13-dihydrodehydrocostuslactone.
2.4 化合物1 和2 對結(jié)腸癌SW620 細胞的細胞毒性結(jié)果顯示,化合物1 和2 對結(jié)腸癌SW620 細胞表現(xiàn)出溫和的細胞毒性(表1)。

表1 轉(zhuǎn)化產(chǎn)物1 和2 對結(jié)腸癌細胞SW620 的細胞毒性
2.5 化合物和結(jié)腸癌SW620 細胞靶點預(yù)測與分子對接結(jié)果 使用GeneCards 數(shù)據(jù)庫和OMIM 數(shù)據(jù)庫獲取到結(jié)腸癌SW620 細胞相關(guān)靶點共960 個;使用Swiss Target Prediction 數(shù)據(jù)庫預(yù)測到化合物1 的靶點有100 個,化合物2 的靶點有57 個;化合物與疾病靶點交集圖如下圖4。分子對接結(jié)果如下表2、3,一般來說結(jié)合能小于-7 kcal/mol 代表結(jié)合作用強烈。取各自結(jié)合能最好的對接組進行對接情況可視化,如下圖5、6。化合物1 與結(jié)腸癌細胞SW620 的交集靶點有26 個,化合物2 與結(jié)腸癌細胞SW620 的交集靶點有16 個。其中化合物1 與靶點蛋白VDR 中的LEU-230、LEU-233 和VAL-234 等多個氨基酸殘基形成多條疏水作用鍵,化合物2 與靶點蛋白CDK1 中的ILE-10、VAL-18 和ALA-31 等多個氨基酸殘基形成多條疏水作用鍵,且兩組結(jié)合能分別為-9.7 kcal/mol 和-8.5 kcal/mol,結(jié)合能優(yōu)異且結(jié)合作用強烈。推斷化合物1 和化合物2 具有抑制結(jié)腸癌細胞SW620的作用。
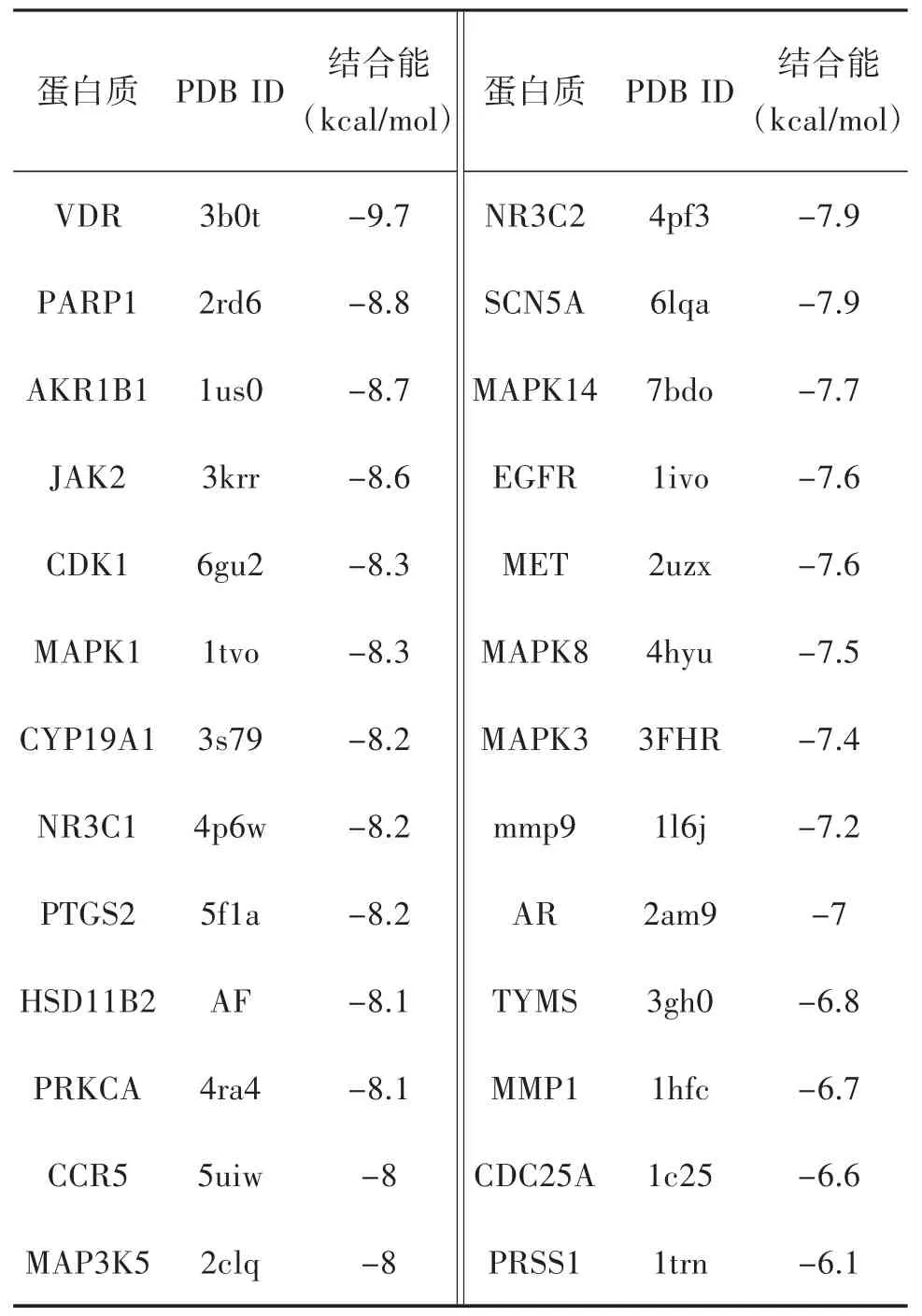
表2 化合物1 與交集靶點結(jié)合能數(shù)據(jù)

表3 化合物2 與交集靶點結(jié)合能數(shù)據(jù)

圖4 化合物1、化合物2 靶點與疾病靶點交集圖
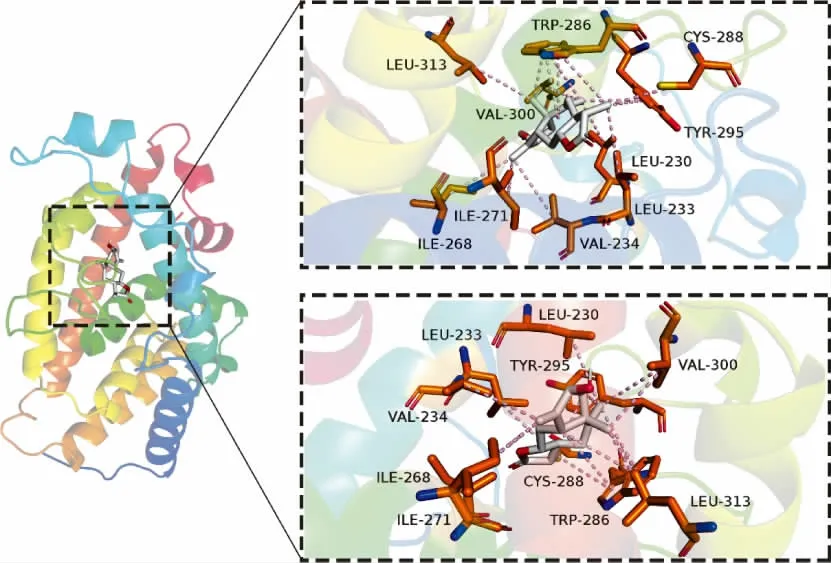
圖5 化合物1 與結(jié)腸癌SW620 細胞的蛋白結(jié)合圖

圖6 化合物2 與結(jié)腸癌SW620 細胞的蛋白結(jié)合圖
3 討論
前期研究了木香烴內(nèi)酯與去氫木香烴內(nèi)酯在Y0液體菌液中的生物轉(zhuǎn)化,第1、3、5、7 天分別對菌液檢測,在第3 天檢測有轉(zhuǎn)化產(chǎn)物生成,但由于木香烴內(nèi)酯的生物轉(zhuǎn)化產(chǎn)率低,未能分離得到轉(zhuǎn)化產(chǎn)物,后期將會繼續(xù)優(yōu)化生物轉(zhuǎn)化條件,以期分離得到轉(zhuǎn)化產(chǎn)物。去氫木香內(nèi)酯在Y0 菌液中轉(zhuǎn)化得到2 個產(chǎn)物,產(chǎn)物1 轉(zhuǎn)化產(chǎn)率為10%,產(chǎn)物2 轉(zhuǎn)化產(chǎn)率為1.4%。作為分子轉(zhuǎn)化的工具,微生物能夠利用酶對傳統(tǒng)化學(xué)合成中的惰性位點進行結(jié)構(gòu)修飾,具有空間選擇性和立體選擇性的特點,能夠?qū)崿F(xiàn)化學(xué)合成難以實現(xiàn)的反應(yīng),解決化學(xué)合成過程繁瑣、污染、成本過高等問題。羥化酶、還原酶及異構(gòu)酶是微生物中常見酶[18],本實驗中通過微生物轉(zhuǎn)化獲得到的2 個產(chǎn)物1 和2 均是去氫木香內(nèi)酯C-3 位發(fā)生羥基化,C-11 發(fā)生還原所得,這表明發(fā)生在C-11 的還原反應(yīng)可能沒有立體選擇的特異性,因此獲得了S、R 兩種構(gòu)型的產(chǎn)物。
轉(zhuǎn)化產(chǎn)物1 和2 對結(jié)腸癌細胞SW620 具有一定的細胞毒性,通過網(wǎng)絡(luò)藥理學(xué)對化合物1、2 靶點預(yù)測,并從GeneCards 數(shù)據(jù)庫和OMIM 數(shù)據(jù)庫獲取結(jié)腸癌SW620 細胞相關(guān)靶點。化合物1 與結(jié)腸癌細胞SW620 的交集靶點有26 個,化合物1 與靶點蛋白維生素D 受體(vitam in D receptor,VDR)結(jié)合能為-9.7 kcal/mol,且 與VDR 中 的LEU-230、LEU-233 和VAL-234 等多個氨基酸殘基形成多條疏水作用鍵,結(jié)合作用強烈。VDR 是一種核轉(zhuǎn)錄因子通過與配體特異結(jié)合調(diào)控多種基因的表達,從而調(diào)節(jié)多種生命活動的進行,在人體各組織細胞中廣泛存在,在結(jié)腸、腎上腺皮質(zhì)、肺等部位的細胞及淋巴細胞中表達量較高[19-20]。Cross 等[21]研究證實,1,25-二羥基維生素D3能有效降低直腸癌、乳腺癌、卵巢癌等多種癌癥的死亡率。近年來大量研究表明:1,25-二羥基維生素D3主要通過依賴VDR 途徑調(diào)節(jié)腫瘤細胞的增殖分化,誘導(dǎo)腫瘤細胞凋亡等,從而抑制腫瘤細胞生長。網(wǎng)絡(luò)藥理與分子對接結(jié)果顯示化合物1 與VDR 有很好的結(jié)合,因此化合物1 有可能通過抑制VDR 發(fā)揮潛在治療結(jié)腸癌的作用。化合物2 與結(jié)腸癌細胞SW620 的交集靶點有16 個,其中與靶點蛋白CDK1 中的ILE-10、VAL-18 和ALA-31 等多個氨基酸殘基形成多條疏水作用鍵,結(jié)合能為-8.5 kcal/mol,結(jié)合作用強烈。細胞周期蛋白依賴性激酶1(cyclin-dependent protein kinase 1,CDK1)是一種高度保守的絲氨酸/蘇氨酸激酶,作為控制細胞周期的起始、進展和終止的主要角色,于細胞周期多個環(huán)節(jié)中均可作為抗癌天然靶標。由于CDK1 在有絲分裂過程的調(diào)控中具有重要作用,已有較多的關(guān)于降低CDK1 活性的物質(zhì)被開發(fā)為抗癌藥物。其中,一些藥物可以通過抑制CDK1 的活性阻滯G2/M 期以達到治療結(jié)直腸癌的目的。例如斑蝥素就是通過抑制CDK1 活性導(dǎo)致結(jié)直腸癌colo 205 細胞中的G2/M 期阻滯和細胞凋亡的[22]。但化合物1、2 抗結(jié)腸癌的活性與可能的作用機制有待進一步的體內(nèi)外實驗來探討。
猜你喜歡
天津醫(yī)科大學(xué)學(xué)報(2019年6期)2019-08-13 07:04:32
西南國防醫(yī)藥(2016年7期)2016-12-01 06:01:15
腹腔鏡外科雜志(2016年10期)2016-06-01 12:10:08
中國衛(wèi)生標準管理(2015年6期)2016-01-14 05:17:12
中國衛(wèi)生標準管理(2015年1期)2016-01-14 03:41:26
中國當代醫(yī)藥(2015年26期)2015-03-01 02:06:57
西南軍醫(yī)(2015年6期)2015-01-23 01:25:50
河南醫(yī)學(xué)研究(2014年3期)2014-02-27 14:51:48
沈陽醫(yī)學(xué)院學(xué)報(2014年1期)2014-02-16 06:19:24
中華介入放射學(xué)電子雜志(2014年1期)2014-02-02 05:24:06
