家畜復雜性狀稀有變異遺傳研究
2023-07-10 06:58:11劉艷萍包阿東梅步俊
安徽農學通報 2023年8期
劉艷萍 包阿東 梅步俊


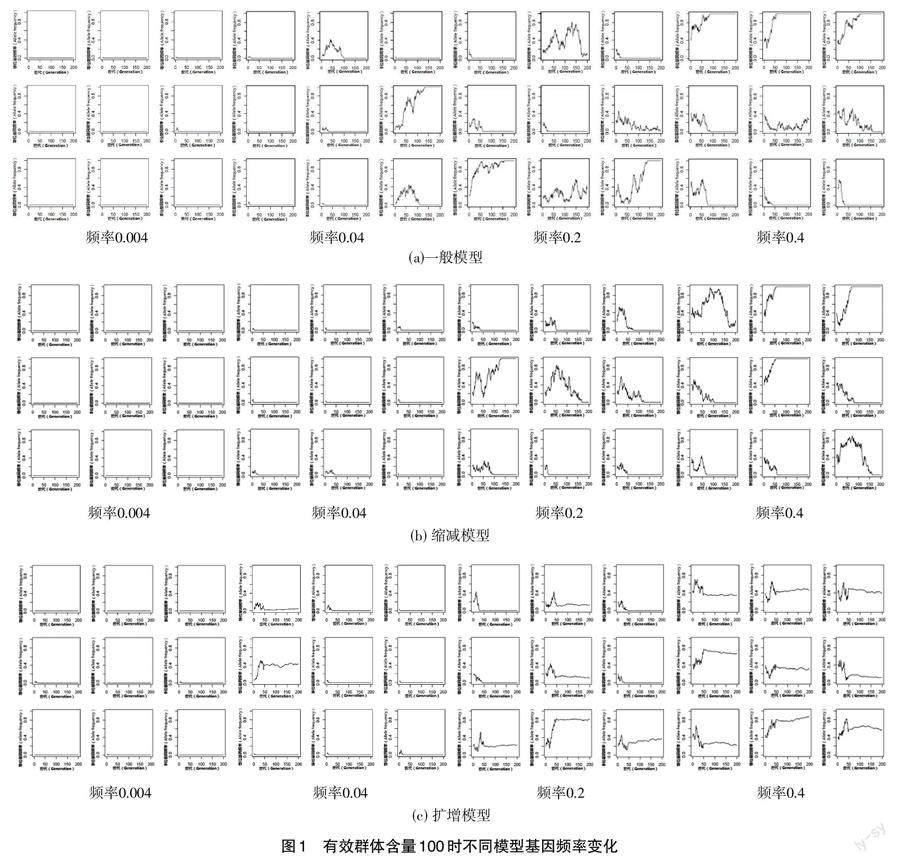
摘要 本研究通過模擬和基因組數據,使用群體遺傳模型來預測稀有變異選擇的預期特征,并比較不同SNP年齡估計方法的結果之間的異同。Wright-Fisher模型框架下,隨著初始基因頻率的增加,基因被固定的概率上升;與有效群體縮減Wright-Fisher模型相比,一般Wright-Fisher模型和有效群體擴增Wright-Fisher模型的基因被固定概率更高,但初始基因頻率較高時,3種模型無明顯差異。稀有變異基因的遺傳方式為共顯性、完全顯性時,稀有變異基因可以在群體中達到很高的頻率或固定,其中完全顯性時基因頻率在群體中達到0.5以上的概率更高;稀有變異基因的遺傳方式為共顯性時,不同有效群體含量隨選擇系數變化的規律與理論預測基本相同。SNP年齡估計依據不同假設條件,往往有不同估計結果。
關鍵詞 復雜性狀;稀有變異;家畜;Wright-Fisher模型;SNP年齡
中圖分類號 S813? ?文獻標識號 A
文章編號 1007-7731(2023)08-0111-08
Study on Inheritance of Rare Variants of Complex Traits in Livestock
LIU Yanping2,3? ?BAO Adong1,2? ?MEI Bujun1,2*
(1Engineering Technology Research Center of Inner Mongolia sheep Genetic Evaluation Method and Application,
Bayannur Inner MongoLia 015000;
2Medical college, Hetao college, Bayannur Inner MongoLia 015000;
3Agriculture department, Hetao college, Bayannur Inner MongoLia 015000)
Abstract This study uses population genetic models to predict the expected characteristics of rare variant selection through simulation and genomic data, and compares the similarities and differences between the results of different SNP age estimation methods. Under the framework of the Wright-Fisher model, with the increase of the initial gene frequency, the probability of the gene being fixed increases; compared with the effective population size contraction Wright-Fisher model, the fixed probability of gene frequency in the general Wright-Fisher model and the effective population expansion Wright-Fisher model is higher, but when the initial gene frequency is higher, there is no significant difference between the those models. When the genetic mode of the rare variant gene is codominance and completely dominant, the rare variant gene can reach a high frequency or fixation in the population. And when the genetic mode is completely dominant, the probability of gene frequency reaching 0.5 or more in the population is higher. When the genetic mode of rare variant genes is codominant, the law of the different effective populations size with the different selection coefficient is basically the same as the theoretical prediction. SNP age estimation methods based on different assumptions often have different estimation results.
Keywords complex traits; rare variants; livestock; Wright-Fisher model; SNP age
全基因組關聯研究(Genome-wide association study,GWAS)方法通過基因芯片技術系統評估常見遺傳變異(通常是SNP,在群體中的最小等位基因頻率MAF>5%)對性狀(疾病)的影響。到目前為止,已經發現有2 000多個SNP與疾病相關,只能解釋小部分的遺傳因素[1]。總體來說,GWAS發現的這些SNP位點對復雜疾病只有中等程度的影響,位點與性狀只有關聯性,沒有因果關系,要想通過關聯關系推導至因果關系仍很困難,要想將這些通過GWAS發現的位點轉化為疾病功能解釋或臨床應用還有很長的路要走。分析低頻位點(0.5% 罕見基因位點一般在基因的編碼區或啟動子區,具有一定的生物學意義,如錯義突變、無義突變、stop loss突變、插入或缺失引起的移碼突變及啟動子區調節轉錄的變異。RVAS是基于功能單元(目前主要是基因,以后有可能是小的信號通路)分析一組位點與疾病的關聯,而GWAS是基于單個位點評估關聯性[3-4]。 突變可能由于遺傳漂變、自然選擇和人工選擇在群體中消失,增加頻率或固定。如果等位基因受到選擇的影響,大的基因組區域也會因遺傳連鎖而受到影響。在強陽性選擇的情況下,基因組區域遺傳變異可能消失,出現“選擇性清除(selective sweep)”[5]。在純化選擇的情況下,當有害等位基因反復從基因座中被清除時,也可以減少連鎖基因組鄰域中的中性突變。這種現象被稱為“背景選擇”。隨著畜禽基因組測序數據的增加,積累了稀有變異數據,這些數據表明畜禽進化過程較預想的復雜,而群體遺傳學研究有助于闡明數量性狀的遺傳機理[6]。本研究通過模擬和基因組數據,使用群體遺傳模型來預測稀有變異選擇的預期特征。 1 材料與方法 1.1 Wright-Fisher模型 本研究分別考慮有效群體含量初始為100和1 000這2種情況,模擬200世代,等位基因初始頻率分別為0.004、0.04、0.2、0.4;分別使用一般Wright-Fisher模型、有效群體縮減Wright-Fisher模型和有效群體擴增Wright-Fisher模型,對于后兩種模型有效群體含量初始為100時,有效群體變動范圍為50;有效群體含量初始為1 000時,有效群體變動范圍為500。 假設一個群體由N個個體組成,[Xt]和[Xt+1]表示[t]和[t]+1世代攜帶等位基因A的染色體數目,分別等于i和j,基因在代際間傳遞可用二項式(Binomial)分布表示: 式中,P為等位基因A的頻率,1-p或q為等位基因a的頻率。 1.2 不同遺傳方式影響 通常根據顯性系數(h)和選擇系數(s)對遺傳模型進行參數化,野生型適應度設置為1,其中基因型AA的適合性為1,基因型Aa的適合性為1+hs,基因型aa的適合性為1+s。h=1為完全顯性,h=0為完全隱性,h=0.5為共顯性。如果種群規模是固定的,要考慮相對適應度,即個體基因型相對于群體的適合度,平均群體適合度[?]為: 1.3 SNP年齡估計方法 20世紀70年代,群體遺傳學家Motoo Kimura和Tomoko Ohta使用等位基因頻率估計等位基因年齡[7]。在隨機交配的大群體中,中性等位基因的年齡估計值為: 式中,p代表等位基因頻率,t1為期望的等位基因年齡,以2N世代為單位。1990年,Jean-Louis Serre等通過分析等位基因內部變異來估計等位基因年齡[8],公式為: 式中,t為世代數,c為重組率,[xt]為t世代時與突變等位基因連鎖的標記頻率,y為與突變等位基因不連鎖的標記頻率。 一般方法是在Serre方法基礎上,計算基因所在區域的LD,計算擴展單倍型雜合性(EHHS, Extended Haplotype Heterozygosity),并在此基礎上設定閾值,計算基因左右翼用于計算年齡的標記長度[9]。本研究使用R軟件SimPhe包內置數據集,比較3種不同SNP年齡估計方法估計結果的異同。 2 結果與分析 2.1 Wright-Fisher模型 Wright-Fisher模型描述了在離散的非重疊世代中有限隨機交配群體的進化。該模型描述了基因座上等位基因頻率隨時間的變化過程。基因頻率受一系列因素的影響,如隨機漂移、突變、遷移、選擇和種群規模的變化。突變、遷移和選擇以確定性方式影響等位基因頻率,被統稱為進化壓力。對有限群體進行隨機抽樣,頻率從一代到下一代變化(遺傳漂移)。突變和遷移會導致抽樣頻率的線性變化,而選擇是一種非線性變化。一般認為,隨著世代推移,等位基因頻率逐漸遠離初始頻率。本研究中(圖1、圖2),當初始基因頻率極低時(0.004),無論種群規模是否發生變化,隨著世代數增加,基因頻率偶爾有微小波動,但在大多數情況下基因始終保持極低頻率或最終從群體中消失;隨著初始基因頻率的增加,基因被固定的概率上升;與有效群體縮減Wright-Fisher模型相比,一般Wright-Fisher模型和有效群體擴增Wright-Fisher模型的基因固定概率更高,但初始基因頻率較高時,3種模型無明顯差異;當有效群體含量由100增加為1 000時,基因更加不容易被固定,且200個世代中,基因頻率最終結果更難預測。 2.2 不同遺傳方式影響 本研究考慮稀有變異3種不同遺傳方式(共顯性、完全顯性和完全隱形),在選擇系數s為0.1時,100個世代基因頻率的變化,共模擬100次(見圖3)。結果顯示,稀有變異基因的遺傳方式為共顯性、完全顯性時,稀有變異基因可以在群體中達到很高的頻率或固定,其中完全顯性時基因頻率在群體中達到0.5以上的概率更高;而遺傳方式為完全隱形時,基因頻率在100個世代內一般不會超過0.4。 本研究用模擬方法比較不同群體含量和選擇系數對稀有變異基因頻率的影響,結果表明,稀有變異基因的遺傳方式為共顯性時,不同有效群體含量隨選擇系數變化的規律與理論預測基本相同;完全顯性時,不同有效群體含量隨選擇系數變化的規律均比理論預測高,而且隨著選擇系數增加,偏差逐漸增大;而完全隱形時,趨勢與完全顯性時相反,且隨著有效群體含量增加,基因頻率隨選擇系數變化規律與理論預測結果的偏差逐漸增大(見圖4)。 2.3 SNP年齡估計方法 等位基因或SNP的年齡可以通過不同拷貝之間的遺傳變異(等位基因內變異)及其頻率來估計。遺傳學界已有估計等位基因年齡的近似方法。只用等位基因頻率也可以估計基因的年齡。基于頻率和等位基因內變異性的估計可以組合以提供更準確的估計,也可以揭示自然選擇的影響是否存在,等位基因年齡的估計取決于對群體歷史和自然選擇的假設。SNP年齡估計依據不同假設條件,往往有不同估計結果,圖5顯示了3種不同估計方法對同一數據估計結果的差異。 3 討論 全基因組關聯研究(GWAS)改變了我們對復雜性狀(例如,體重指數BMI,血壓和血脂)遺傳基礎的理解,以及對常見1型和2型糖尿病、冠狀動脈疾病等疾病的認識。盡管GWAS成功地鑒定了與這些復雜性狀相關的基因組區域,但是僅能解釋遺傳力的一小部分。GWAS的一個重要局限性在于,通常以常見變異關聯信號為理論基礎,每個信號僅對性狀產生一定的效應。因此,人們越來越傾向“缺失遺傳力”由罕見的遺傳變異或低頻變異(通常定義為MAF<1%)引起[10]。 全基因組測序技術是研究稀有遺傳變異的金標準。傳統GWAS方法分析群體中頻率低于1%變異的功效有限。但下一代測序技術和新型分析技術的發展,稀有遺傳變異在許多復雜性狀中都發揮了作用,包括克羅恩病中的NOD2、1型糖尿病的IFIH1,病態竇房結綜合征中的MYH6和調節空腹血糖水平的G6PC2。正在進行的基于人類群體的全基因組測序計劃,例如千人基因組和UK10K項目,正在為跨群體研究稀有遺傳變異的分布和特征提供了基礎,通過改進的填補技術,可以對數以千萬計具有復雜性狀的變異進行關聯研究。但DNA芯片技術通常對于常見SNP變異最有效,能夠達到99.5%以上的準確度,而對于低頻和稀有變異(次要等位基因頻率<5%)則準確性不高[11]。隨著針對稀有變異(例如Exome Chip和MetaboChip)芯片的廣泛使用,已經發布了精確識別稀有變異的新方法。 Affymetrix和Illumina兩家公司有專門針對低頻變異(次等位基因頻率<5%)的基因芯片,可以用于遺傳學關聯研究。現有用于常見SNP的芯片基因型分型算法(如MPAM、DM、RLMM、GEL、BRLMM、CRLMM、CHIAMO等)不適用于稀有變體,因為這些方法在對數據進行聚類時會假定存在3個基因型類,而稀有變異則不適用這種假設。例如,對于處于哈代溫伯格平衡的次等位基因頻率<1%的變異,則需要對100 000個樣本進行基因分型,以期望每個基因型類別至少有10個信號[12]。另外,很難對稀有變異的算法進行基準校對,因為大多數樣品(包括HapMap樣品)的稀有變體的檢測率都與常見變異不在同一數量級。使用家系數據也不一定能解決這個問題。如果未檢測到稀有基因型,并且家系中的每個個體都被稱為常見等位基因純合子,且不會發生孟德爾錯誤。類似地,因為無法判斷稀有變異等位基因基因型的正確識別率,比較算法結果與已知基因型的總體一致性幾乎沒用。例如,對于MAF=0.1%的SNP,計算所有位點常見等位基因純合子的算法有99.8%的正確率[13]。因此,對于稀有變異,找到合適的比較數據集和評估算法的準確性比常見變異更具挑戰性。 SNP是最常見的遺傳突變,也可能是研究最多的遺傳變異。SNP檢測的原理較簡單,在讀段正確比對到參考序列以后,與參考序列比對不上的堿基就是SNP。但有許多因素會導致出現“假SNP”。常見導致“假SNP”的5種原因:①測序錯誤,測序儀本身導致“假SNP”的出現,讀段中存在不正確的堿基,有時錯誤的堿基會以低質量序列打分數被反映出來;②PCR錯誤,PCR擴增過程中發生復制錯誤,導致出現堿基錯誤的讀段,多次對同一PCR片段重復測序可能會加劇這種情況的出現;③污染,生物樣品包含來自另一個樣品或物種(例如細菌或病毒)的少量DNA;④比對錯誤,如將讀段比對到錯誤的基因組位置,則可能導致明顯的堿基不匹配;⑤存在未知的插入或缺失,如果變異附近存在未知的變異(例如大片段的插入或缺失),則位于該變異側翼的讀段可能比對不正確,從而產生SNP,這種錯誤較難發現和矯正[14]。可以通過以下3種方式判斷SNP檢測率是否有錯誤:①如在人類中,計算SNP的發生率是否約為每1 000個堿基1個;②轉換/顛換比(Ts/Tv),已知自然發生轉換形成的SNP(C→T,T→C,A→G,G→A)的速率顯著高于顛換形成SNP的速率,正常情況下該比例應高于2;③等位基因頻率:實際SNP數據應當在0.5和1.0等位基因頻率處存在2個峰值,如果在0.5和1.0處沒有明顯的峰值,則SNP檢測有問題[15]。 稀有變異的質控主要是去除錯誤率較高的變異和個體,以免產生虛假的關聯信號。通常樣本水平的質控優先于變異水平的質控,以確保質量不合格的樣本不影響后續分析。樣品混合是造成測序錯誤的主要原因,可能發生在整個采樣及測序過程中。通常可以使用“DNA指紋”技術檢測樣品登記性別和測序標記估計“性別”一致性,或將測序結果與以前個體測序基因型相比較來判斷樣品是否有混合。可以使用Sequenom測序平臺測得的幾十個常見多態性位點作為“DNA指紋”[16, 17]。基因型錯誤可以利用同源一致性原理進行校正。 等位基因年齡(或突變年齡)指等位基因首次突變出現以來經過的時間。估計某個等位基因出現的時間可以推斷物種遷徙,性狀和自然選擇的模式。估計等位基因年齡主要依據2種方式:①群體中等位基因的頻率;②等位基因不同拷貝內發生的遺傳變異,也稱為等位基因內變異[18]。將2種方式聯合使用可以提高等位基因年齡估計的準確性,有時也可以提供選擇信號是否存在的信息。基于頻率估計等位基因年齡的基本假設:在沒有選擇的情況下,高頻等位基因比低頻等位基因更古老。在有正向選擇的情況下,由于自然選擇、基因流、遺傳漂移和突變等因素可能使等位基因頻率增長較快。基于等位基因內部變異來估計等位基因年齡的基本假設,即每一世代重組均會破壞連鎖不平衡,也會產生連鎖的新變異。基于等位基因內變異估計等位基因年齡可以使用溯祖理論(coalescent theory),分析年代久遠的歷史突變時可以通過重建基因樹并確定樹根的方式來推斷等位基因的年齡,而對于新近突變,群體遺傳學使用突變,重組率和統計學模型來估計等位基因年齡[19]。 等位基因年齡估計的例子:囊性纖維化(Cystic fibrosis)、艾滋病抗性等位基因(AIDS-resistance allele,CCR5)、乳糖酶持久性(Lactase persistence)。最近,Albers和McVean[20]提出了一種非參數方法,使用基于聚結的突變和重組模型來估計等位基因的年齡。具體而言,該方法將時間推斷為成百上千的染色體序列(單倍型)對之間的最新共同祖先(TMRCA),然后使用復合似然方法將該信息組合起來,以獲得單個基因座突變時間的估計值。使用來自1 000個基因組計劃和西蒙斯基因組多樣性計劃的數據,該方法應用于人類基因組中的1 600萬個變體,以生成變體年齡圖集[21]。 4 作者貢獻 梅步俊負責文章的設計、撰寫及程序編寫,是試驗設計和試驗研究的執行人;包阿東和劉艷萍負責編寫部分程序和資料整理,參與部分試驗設計、試驗結果分析。全體作者都閱讀并同意最終的文本。 5 致謝 感謝華中農業大學姜勛平教授課題組在研究過程中提供的幫助。本研究受國家自然科學基金項目(31760660);內蒙古自治區自然科學基金項目(2019MS03092);內蒙古自治區肉羊遺傳評估方法與應用工程技術研究中心;巴彥淖爾市科技創新基金項目;巴彥淖爾市科技計劃項目(BKZ2016);內蒙古自治區科技計劃項目(2020GG0201)等項目的資助。 6 參考文獻 [1] BURKETT K M,MCNENEY B,GRAHAM J,et al. Using gene geneal-ogies to detect rare variants associated with complex traits[J]. Hum Hered.,2014,78(3-4):117-130. [2] CHEN G,YUAN A,ZHOU Y,et al. Simultaneous analysis of common and rare variants in complex traits:Application to SNPs (SCARVAsnp)[J]. Bioinform Biol Insights,2012(6):177-185. [3] FENG T,ELSTON R C,ZHU X. Detecting rare and common variants for complex traits:sibpair and odds ratio weighted sum statistics (SPWSS,ORWSS)[J]. Genet Epidemiol.,2011,35(5):398-409. [4] OSCAR G R,DAETWYLER H D,MACLEOD I M,et al. Rare variants in transcript and potential regulatory reg-ions explain a small percentage of the missing heritability of complex traits in cattle[J]. PLoS One,2015,10(12):e0143945. [5] JOUAN L,GAUTHIER J,DION P A,et al. Rare variants in complex traits:novel identification strategies and the role of de novo mutations[J]. Hum Hered.,2012,74(3-4):215-25. [6] KARUNARATHNA C B,GRAHAM J. Using gene genealogies to localize rare variants associated with complex traits in diploid populations[J]. Hum Hered.,2018,83(1):30-39. [7] SLATKIN M,RANNALA B. Estimating allele age[J]. Annu Rev Genomics Hum Genet.,2000(1):225-49. [8] Malaspinas,Anna-Sapfo. Estimating allele age and selection coefficient from time-serial data[J]. Genetics,2012,192(2):599-607. [9] GANDOLFO,LUKE,C,et al. Dating rare mutations from small samples with dense marker data[J]. Genetics,2014,197(4):1315-1327. [10] KIM S,LEE K,SUN H. Statistical selection strategy for risk and protective rare variants associated with complex traits[J]. J Comput Biol.,2015,22(11):1034-1043. [11] LETTRE,G. Rare and low-frequency variants in human common diseases and other com-plex traits[J]. J Med Genet.,2014,51(11):705-714. [12] LI B,LIU D J,LEAL S M. Identifying rare variants associated with complex traits via sequencing[J]. Curr Protoc Hum Genet.,2013(1):1-26. [13] LU T H,AUSTIN E,BONNER A,et al. Applications of machine learning and data mining methods to detect associations of rare and common variants with complex traits[J]. Genet Epidemiol.,2014,38(1):81-85. [14] MR MUNAF?,FLINT J. Common or rare variants for complex traits?[J]. Biol Psychiatry,2014,75(10):752-753. [15] KALLIOPE P,IOANNA T,ELEFTHERIA Z. In search of low-frequency and rare variants affecting complex traits[J]. Hum Mol Genet.,2013,22(R1):16-21. [16] RITCHIE G R S,FLICEK P. Functional annotation of rare genetic variants,in assessing rare variation in complex traits:design and analysis of genetic studies[M]. E. Zeggini and A. Morris,Editors. New York(NY):2015 57-70. [17] RUDRA P,BROADAWAY K A,WARE E B,et al. Testing cross-phenotype effects of rare variants in longitudinal stud-ies of complex traits[J]. Genet Epidemiol.,2018,42(4):320-332. [18] DUBON M,PEDROSA V,FEITOSA F,et al. Identification of novel candidate genes for age at first calving in Nellore cows using a SNP chip specifically developed for Bos taurus indicus cattle[J]. Theriogenology,2021,173:156-162. [19] SCHMIDT N,K SCH?CKER,KRAUSE I,et al. Genome-wide SNP typ-ing of ancient DNA:Determination of hair and eye color of Bronze Age humans from their skeletal remains[J]. Am J Phys Anthropol.,2020,172(1):99-109. [20] HAMANN L,SZWED M,MOSSAKOWSKA M,et al. First evidence for STING SNP R293Q being protective regarding obesity-associated cardiovascular disease in age-advanced subjects-a cohort study[J]. Immun Ageing,2020,17:7. [21] SINGH A,BABYAK M A,SIMS M,et al. Evaluating the precision of EBF1 SNP x stress interaction association:sex,race,and age differences in a big harmonized data set of 28,026 participants[J]. Transl Psychiatry,2020,10(1):351. (責編:何 艷) 基金項目 國家自然科學基金項目(31760660);內蒙古自治區自然科學基金項目(2019MS03092);巴彥淖爾市科技計劃項目(BKZ2016);內蒙古自治區科技計劃項目(2020GG0201)。 作者簡介 劉燕萍(1969—),女,副教授。研究方向:動物胚胎移植及家畜的保種和育種。 收稿日期 2021-12-06
