玄參藥材中多類型成分的定量分析方法建立及其在脈絡(luò)寧口服液中的應(yīng)用
2023-06-08 12:21:22徐思易譚亞杰唐浩竣譚寧華
中草藥 2023年11期
關(guān)鍵詞:質(zhì)量
徐思易,譚亞杰,唐浩竣,李 劍,2*,譚寧華*
1. 中國藥科大學中藥學院,江蘇 南京 211198
2. 金陵藥業(yè)股份有限公司,江蘇 南京 210009
脈絡(luò)寧口服液(Mailuoning Oral Liquid,MOL)為江蘇省中醫(yī)藥研究所顧亞夫教授課題組在臨床實踐的基礎(chǔ)上所研制,由金陵藥業(yè)股份有限公司獨家生產(chǎn),具有清熱養(yǎng)陰、活血化瘀的功效。處方由君藥牛膝、臣藥玄參、佐藥石斛、使藥金銀花和山銀花5 味中藥材組成,臨床上常用于治療血栓閉塞性脈管炎、靜脈血栓形成、動脈硬化性閉塞癥、腦血栓形成及其后遺癥。臣藥玄參來源于玄參科玄參屬植物玄參ScrophularianingpoensisHemsl.的干燥根[1]。研究表明,玄參提取液具有保護腦缺血損傷[2-4]、抗動脈粥樣硬化[5]和免疫調(diào)節(jié)[6]等藥理活性。因此,玄參的化學成分可能為MOL 發(fā)揮藥效的關(guān)鍵成分,其藥材質(zhì)量優(yōu)劣將直接影響其臨床療效。目前玄參的成分分析和含量測定方法主要用于藥材的產(chǎn)地差異[7]、規(guī)格等級[8-9]、加工方法[10-13]和藥用部位[14-15]等的質(zhì)量評價,其成分分析主要集中在一類或少數(shù)幾個指標性成分考察。迄今為止,尚未見同時定量玄參中4 類有效成分,如環(huán)烯醚萜苷、苯丙素苷、有機酸及核苷類等,用于評價不同產(chǎn)地藥材質(zhì)量的相關(guān)報道。
基于玄參藥材成分復(fù)雜且部分定量的成分含量低,本研究采用超高效液相色譜-串聯(lián)三重四極桿質(zhì)譜(UPLC-QqQ-MS/MS)方法在多反應(yīng)監(jiān)測(multiple reaction monitoring,MRM)模式下,首次建立玄參中環(huán)烯醚萜苷、苯丙素苷、有機酸及核苷4 類有效成分的分析方法,并將此方法首次應(yīng)用于MOL,實現(xiàn)了玄參藥材中25 個成分和MOL 中15個成分的同時定量。結(jié)合化學計量學方法對不同產(chǎn)地玄參藥材和不同批次MOL 進行質(zhì)量評價,旨在為MOL 組方中玄參藥材的選擇及其復(fù)方的質(zhì)量控制提供更多科學依據(jù)。
1 儀器與材料
1.1 儀器
Waters Acquity?UPLC H-Class 超高效液相色譜儀,Waters XEVO?TQD system 型三重四極桿質(zhì)譜儀,美國沃特世公司;KQ-200KDE 型數(shù)控超聲清洗器,昆山市超聲儀器有限公司;XS105 型十萬分之一電子天平,梅特勒-托利多公司。
1.2 材料
對照品腺苷(R1,批號N24D11W135689)和對甲氧基肉桂酸(R22,批號A31J10L94348)購自上海源葉生物科技有限公司;2′-脫氧腺苷(R2,批號AF21081054)、鳥苷(R3,批號AF20073153)和地黃苷(R18,批號AF21102904)購自成都埃法生物科技有限公司;桃葉珊瑚苷(R4,批號DSTDT000401)、哈巴苷(R5,批號DST210329-059)、原兒茶酸(R6,批號DSTDY008101)、對羥基苯甲酸(R7,批號DSTDD011401)、對香豆酸(R9,批號DSTDD005701)、異毛蕊花糖苷(R13,批號DST210601-060)、安格洛苷 C(R14,批號DST211010-009)、哈巴俄苷(R20,批號DST220312-058)和肉桂酸(R21,批號DSTDG016801)購自成都德斯特生物科技有限公司;咖啡酸(R8,批號PRF20070342)購自成都普瑞法科技開發(fā)有限公司;毛蕊花糖苷(R10,批號20122101)購自成都普菲徳生物科技有限公司;阿魏酸(R12,批號110773-201915)購自中國食品藥品檢定研究院;斬劍龍苷A(R11)、肉蓯蓉苷C(R15)、8-O-對香豆酰基哈巴苷(R16)、1-(3-hydroxy-4-methoxyphenyl) ethyl-3-α-L-rhamnopyranosyl-6-caffeoyl-β-D-glucopyranoside(R17)、異地黃苷(R19)、scrophuloside A4(R23)、scrophuloside B4(R24)和keolzioside(R25)由本實驗室提供。以上購買的對照品質(zhì)量分數(shù)≥98%,自制的對照品質(zhì)量分數(shù)≥95%。乙腈(德國Merck公司)和甲酸[賽默飛世爾科技(中國)有限公司]均為LC-MS 級,水為Milli-Q 系統(tǒng)制備的超純水,其余試劑為分析純。
重慶市(S1、S2)、貴州省(S3~S5)、浙江省(S6~S8)與河南省(S9、S10)產(chǎn)地的玄參藥材由金陵藥業(yè)股份有限公司提供;河北省(S11~S13)、湖北省(S14~S16)與安徽省(S17~S19)產(chǎn)地的玄參藥材購買于亳州藥材市場。玄參藥材由中國藥科大學中藥學院譚寧華教授鑒定為玄參科玄參屬植物玄參S.ningpoensisHemsl.的干燥根。53 批MOL由金陵藥業(yè)股份有限公司提供,樣品信息見表1。

表1 53 批MOL 樣品信息Table 1 Sample information of 53 batches of MOL
2 方法與結(jié)果
2.1 色譜與質(zhì)譜條件
2.1.1 色譜條件 采用Waters Acquity UPLC?HSS T3 色譜柱(100 mm×2.1 mm,1.8 μm);流動相為0.1%甲酸水溶液和乙腈,梯度洗脫:0~3.0 min,0~10%乙腈;3.0~5.0 min,10%~20%乙腈;5.0~12.0 min,20%~30%乙腈;12.0~13.0 min,30%~100%乙腈;13.0~15.0 min,100%乙腈;體積流量0.3 mL/min;柱溫30 ℃;進樣量2 μL。
2.1.2 質(zhì)譜條件 電噴霧(electrospray ionization,ESI)離子源,電離模式ESI+/ESI-,掃描方式采用多反應(yīng)監(jiān)測(multiple reaction monitoring,MRM)模式,脫溶劑氣體積流量650 L/h;脫溶劑氣溫度650 ℃;離子源溫度150 ℃;錐孔體積流量50 L/h;毛細管電壓2.0 kV;25 個成分優(yōu)化后的質(zhì)譜參數(shù)見表2。各成分在混合對照品溶液和樣品溶液中的總離子流圖見圖1,疊加后的MRM 色譜圖見圖2。
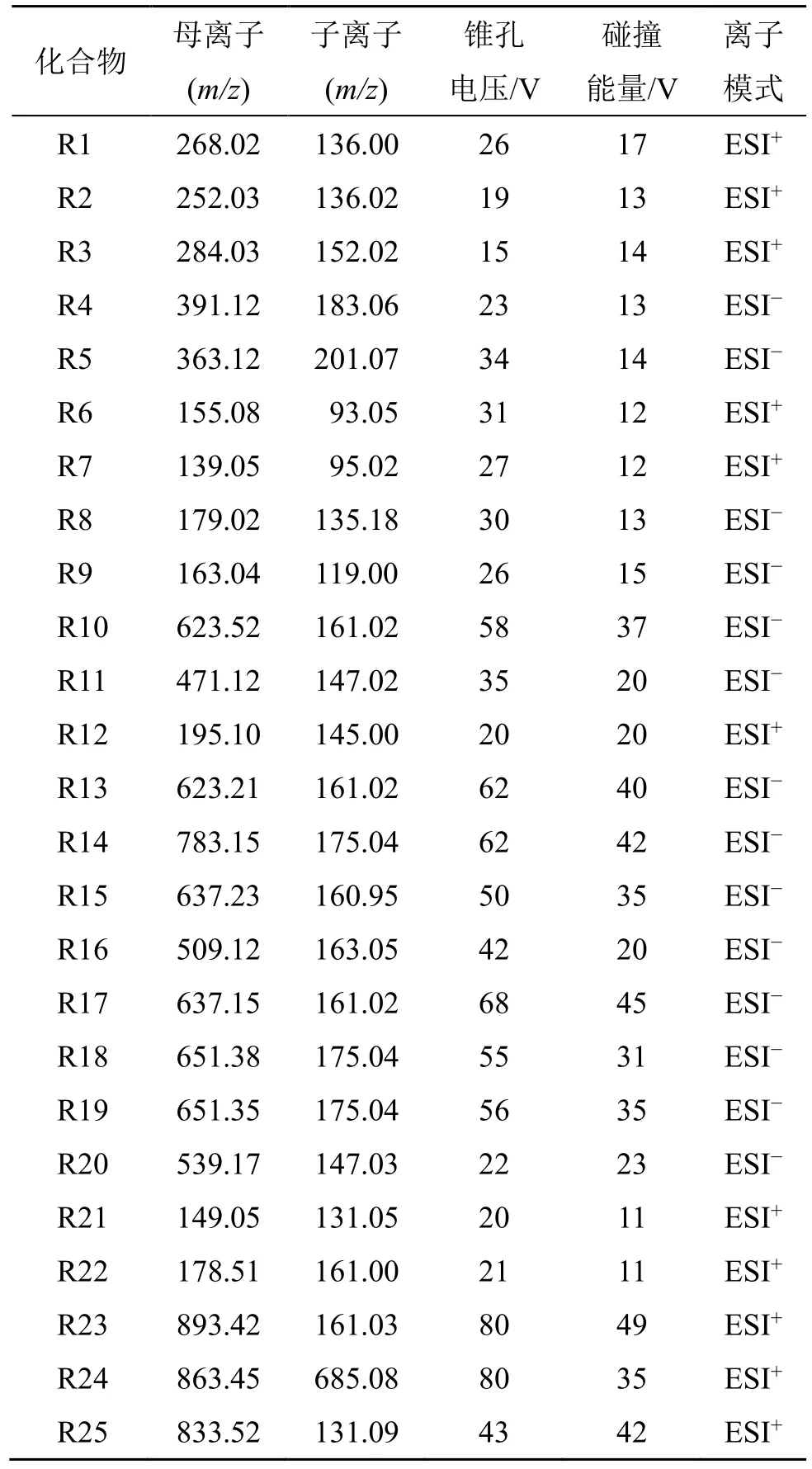
表2 25 種成分的MS/MS 參數(shù)Table 2 MS/MS parameters of 25 compounds
2.2 對照品溶液的制備
分別精密稱定25 個對照品適量,加70%甲醇制成各對照品儲備液。取各對照品儲備液適量,制成R1~R25 質(zhì)量濃度分別為20.00、0.084、4.70、40.00、388.80、12.90、5.50、31.20、10.30、72.00、20.00、10.10、18.56、320.00、16.00、10.80、10.50、10.00、5.25、153.60、63.60、5.05、5.40、11.30、5.30 μg/mL 的混合對照品溶液,置于4 ℃下保存,備用。
2.3 供試品溶液的制備
2.3.1 玄參藥材供試品溶液 取玄參藥材粉末(過50 目篩)約0.5 g,精密稱定,置100 mL 具塞錐形瓶中,精密移取70%甲醇30 mL,密塞,稱量,超聲處理40 min(200 W、40 ℃),放冷,稱量,用70%甲醇補足減失的質(zhì)量,搖勻,過0.22 μm 有機濾膜,取續(xù)濾液,即得玄參藥材供試品溶液。
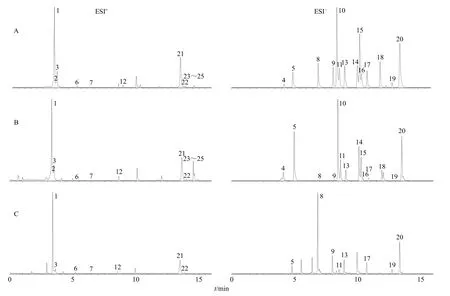
圖1 化合物R1~R25 (1~25) 在混合對照品 (A)、玄參藥材 (B) 和MOL (C) 中的總離子流圖Fig. 1 Total ion chromatograms of compounds R1—R25 (1—25) in mixed standard solutions (A), Scrophulariae Radix (B) and MOL (C)
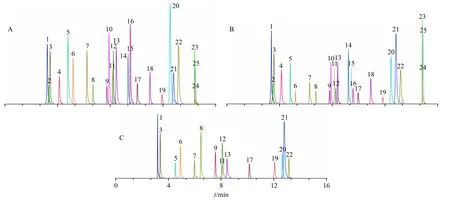
圖2 化合物R1~R25 (1~25) 在混合對照品 (A)、玄參藥材 (B) 和MOL (C) 中的MRM 色譜圖Fig. 2 MRM chromatograms of compounds R1—R25 (1—25) in mixed standard solutions (A), Scrophulariae Radix (B) and MOL (C)
2.3.2 MOL 供試品溶液 精密吸取MOL 1 mL,置10 mL 量瓶中,加水定容至刻度線,搖勻,過0.22 μm 有機濾膜,取續(xù)濾液,即得MOL 供試品溶液。
2.4 方法學考察
2.4.1 線性關(guān)系、檢測限與定量限考察 取“2.2”項下混合對照品溶液,按2 倍逐級稀釋得HB-1~HB-12,于上述“2.1”項下的條件下測定。以對照品質(zhì)量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸分析,繪制標準曲線(至少6 個質(zhì)量濃度點),以信噪比S/N=3 和S/N=10 分別計算檢測限和定量限。結(jié)果見表3,表明25 個化合物在線性范圍內(nèi)線性關(guān)系良好。

表3 25 個化合物的線性關(guān)系考察結(jié)果Table 3 Linear range investigation of 25 compounds
2.4.2 精密度考察 取同一混合對照品溶液,按照“2.1”項的條件連續(xù)進樣6 次,計算各成分峰面積的RSD,得日內(nèi)精密度;連續(xù)3 d 精密吸取同一混合對照品溶液,重復(fù)進樣6 次,計算各成分峰面積的RSD,得日間精密度。結(jié)果表明,R1~R25 日內(nèi)的RSD 分別為1.84%、2.08%、0.83%、1.61%、1.22%、1.90%、2.65%、1.93%、4.08%、2.47%、2.45%、1.77%、1.97%、1.39%、2.46%、2.09%、2.48%、3.07%、2.40%、3.67%、1.50%、5.65%、4.41%、5.99%、4.38%,日間的RSD 分別為3.77%、2.66%、4.26%、5.74%、7.42%、3.74%、3.72%、1.38%、7.33%、7.09%、10.41%、6.08%、7.11%、5.42%、6.78%、5.38%、6.03%、7.34%、5.44%、4.45%、4.35%、7.76%、4.26%、4.98%、2.12%,表明儀器精密度良好。
2.4.3 重復(fù)性考察 按“2.3”項下方法分別平行制備6 份玄參藥材(S1)和MOL(M26)的供試品溶液,計算6 份玄參提取液中各成分峰面積/取樣量的RSD 和MOL 樣品中各成分峰面積的RSD。結(jié)果顯示,玄參藥材中R1~R25 的RSD 分別為1.93%、4.29%、2.16%、2.26%、1.02%、4.22%、3.16%、4.76%、4.90%、0.95%、1.66%、3.24%、2.40%、2.40%、1.09%、2.43%、3.14%、2.96%、5.27%、3.57%、0.93%、1.25%、2.75%、3.77%、2.22%,MOL 中R1、R3、R5~R9、R11~R13、R17、R19~R22 的RSD 分別為1.00%、2.58%、3.01%、2.45%、3.05%、0.66%、1.51%、2.67%、1.87%、2.92%、2.14%、7.71%、7.57%、2.75%、5.14%,表明該方法重復(fù)性良好。
2.4.4 穩(wěn)定性考察 取同一份供試品溶液,按照“2.1”項下條件分別于0、2、4、8、12、24 h 進樣分析,計算各成分峰面積的RSD。結(jié)果顯示,玄參藥材樣品(S1)中R1~R25 的RSD 分別為2.15%、6.13%、3.23%、4.88%、2.75%、8.82%、6.94%、3.91%、3.50%、2.24%、4.02%、5.90%、5.23%、4.10%、4.47%、5.45%、4.08%、5.91%、7.02%、1.99%、2.82%、6.03%、5.69%、9.91%、2.04%,MOL(M26)中R1、R3、R5~R9、R11~R13、R17、R19~R22 的RSD 分別為1.70%、1.26%、2.88%、0.98%、2.40%、0.94%、1.99%、5.01%、0.91%、2.21%、2.56%、3.00%、3.43%、1.39%、4.82%,結(jié)果表明,供試品溶液在24 h 內(nèi)穩(wěn)定性良好。
2.4.5 加樣回收率考察 分別精密稱定已測定指標成分含量的0.25 g 玄參藥材樣品(S1)9 份和精密吸取已測定指標成分含量的0.5 mL MOL 樣品(M26)9 份,每3 份1 組。分別按照低、中、高3個水平(50%、100%、150%)精密加入對照品溶液,按“2.3”項下方法制備供試品溶液,按“2.1”項下的條件進樣分析,計算加樣回收率。結(jié)果顯示,玄參藥材中 R1~R25 的平均加樣回收率分別為97.10%、91.82%、112.22%、84.80%、84.75%、87.90%、108.27%、107.69%、98.92%、101.40%、98.51%、104.42%、104.66%、104.74%、89.82%、111.28%、107.02%、100.79%、86.35%、110.01%、101.65%、107.10%、104.04%、107.72%、109.49%,RSD 分別為5.52%、3.54%、7.20%、4.12%、5.09%、5.51%、7.84%、6.00%、6.66%、1.56%、1.38%、7.02%、2.66%、3.87%、3.13%、3.32%、3.55%、2.57%、6.54%、9.48%、1.75%、6.14%、6.91%、9.01%、4.11%;MOL 樣品中R1、R3、R5~R9、R11~R13、R17、R19~R22的平均加樣回收率分別為 106.20%、94.06%、111.92%、97.42%、105.71%、100.64%、86.09%、105.02%、100.34%、101.61%、106.47%、92.74%、102.90%、105.82%、93.28%,RSD 分別為4.28%、3.44%、8.52%、4.38%、4.34%、2.61%、7.47%、3.35%、8.33%、7.39%、5.34%、3.83%、7.48%、3.09%、4.32%;結(jié)果表明該方法準確度較高。
2.5 樣品含量測定
取不同批次玄參藥材樣品和MOL 樣品,按“2.3”項下方法制備供試品溶液,按“2.1”項下條件進樣分析。根據(jù)回歸方程計算各化合物的含量,各批次樣品含量見表4、5,疊加示意圖如圖3、4 所示,不同產(chǎn)地玄參藥材和不同年份的MOL 中各成分總含量有明顯差異。玄參藥材中25 種成分的總含量以華北地區(qū)(河北省)為最高,浙江省次之。MOL 2021 和2022 年樣品中15 種成分的總含量整體高于2019 年和2020 年的樣品。
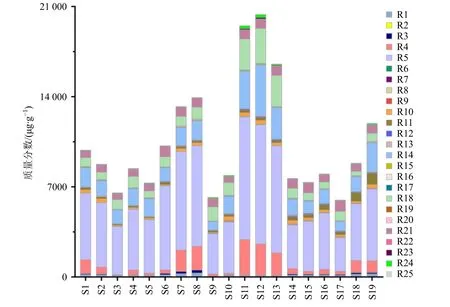
圖3 19 個批次玄參藥材樣品中25 種成分含量的疊加示意圖Fig. 3 Overlay diagram of contents of 25 compounds in 19 batches of Scrophulariae Radix samples

圖4 53 個批次MOL 樣品中15 種成分含量的疊加示意圖Fig. 4 Overlay diagram of contents of 15 compounds in 53 batches of MOL samples
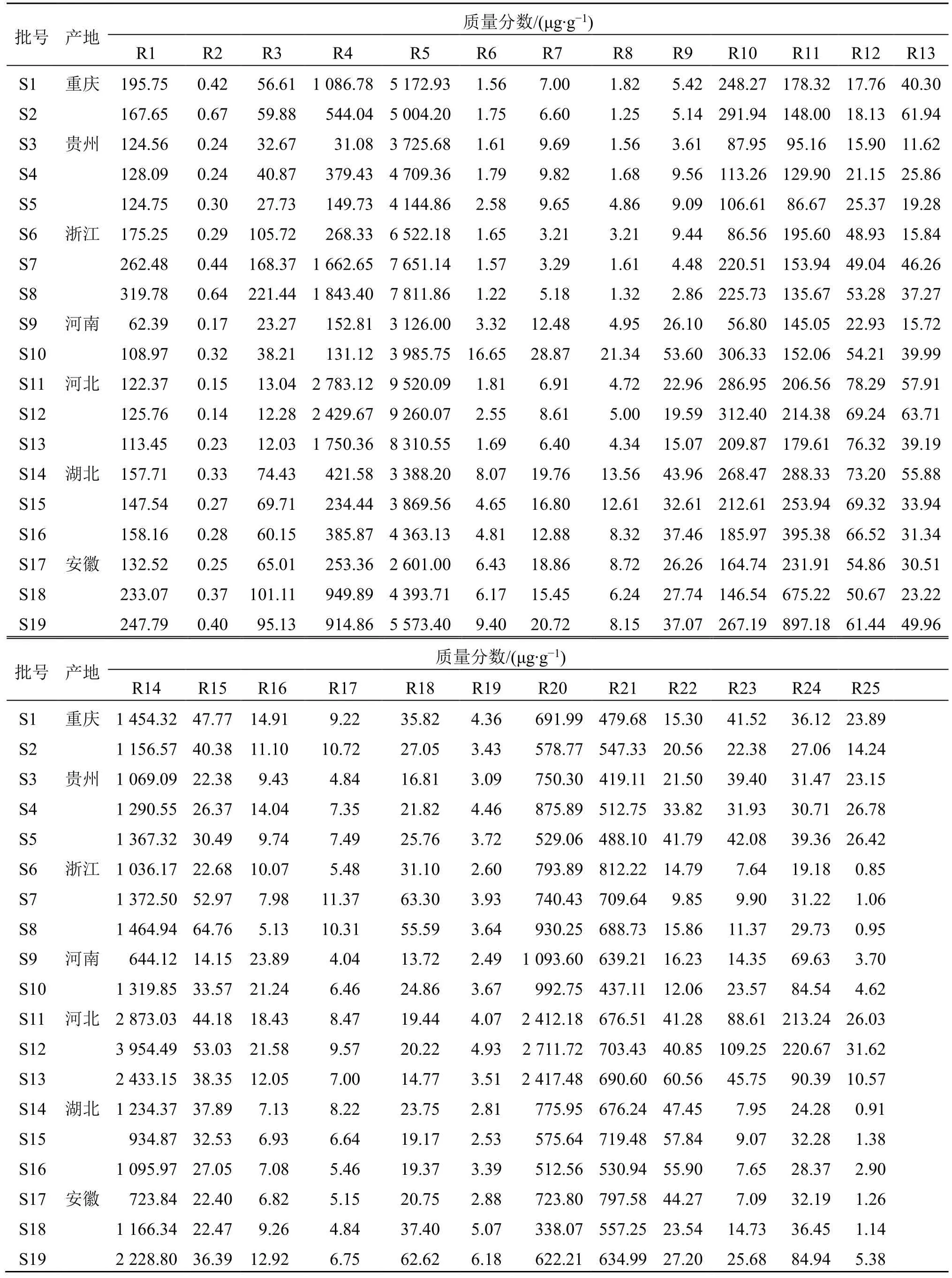
表4 19 個批次玄參藥材樣品中25 種成分的含量Table 4 Contents of 25 compounds in 19 batches of Scrophulariae Radix samples

表5 53 個批次MOL 樣品中15 種成分的含量Table 5 Contents of 15 compounds in 53 batches of MOL samples

續(xù)表5
2.6 化學計量學分析
2.6.1 聚類分析(cluster analysis,CA) 采用HemI軟件對不同批次樣品的化學成分含量進行聚類熱圖分析,數(shù)據(jù)進行歸一化取對數(shù)值后再進行雙向聚類,結(jié)果見圖5 和6。19 批玄參藥材樣品按地域被分為4 類:西南地區(qū)(重慶S1、S2,貴州S3~S5)為第1 類,華東沿海地區(qū)(浙江S6~S8)為第2 類,華東內(nèi)陸和華中地區(qū)(安徽S17~S19,河南S9、S10,湖北S14~S16)為第3 類,華北地區(qū)(河北S11~S13)為第4 類,說明玄參藥材樣品中各成分的含量可能與其生長環(huán)境相關(guān)。53 批MOL 樣品被分為2類:過期樣品(M1~M11)為第1 類,含量相對較低;未過期樣品(M12~M53)為第2 類,含量相對均勻,表明MOL 在有效期內(nèi)各成分含量較接近,過期樣品中各成分含量下降。此外,2 類中不同年份的樣品又可再聚成一小類,可能是當年使用的藥材質(zhì)量相對穩(wěn)定使其含量更相近。
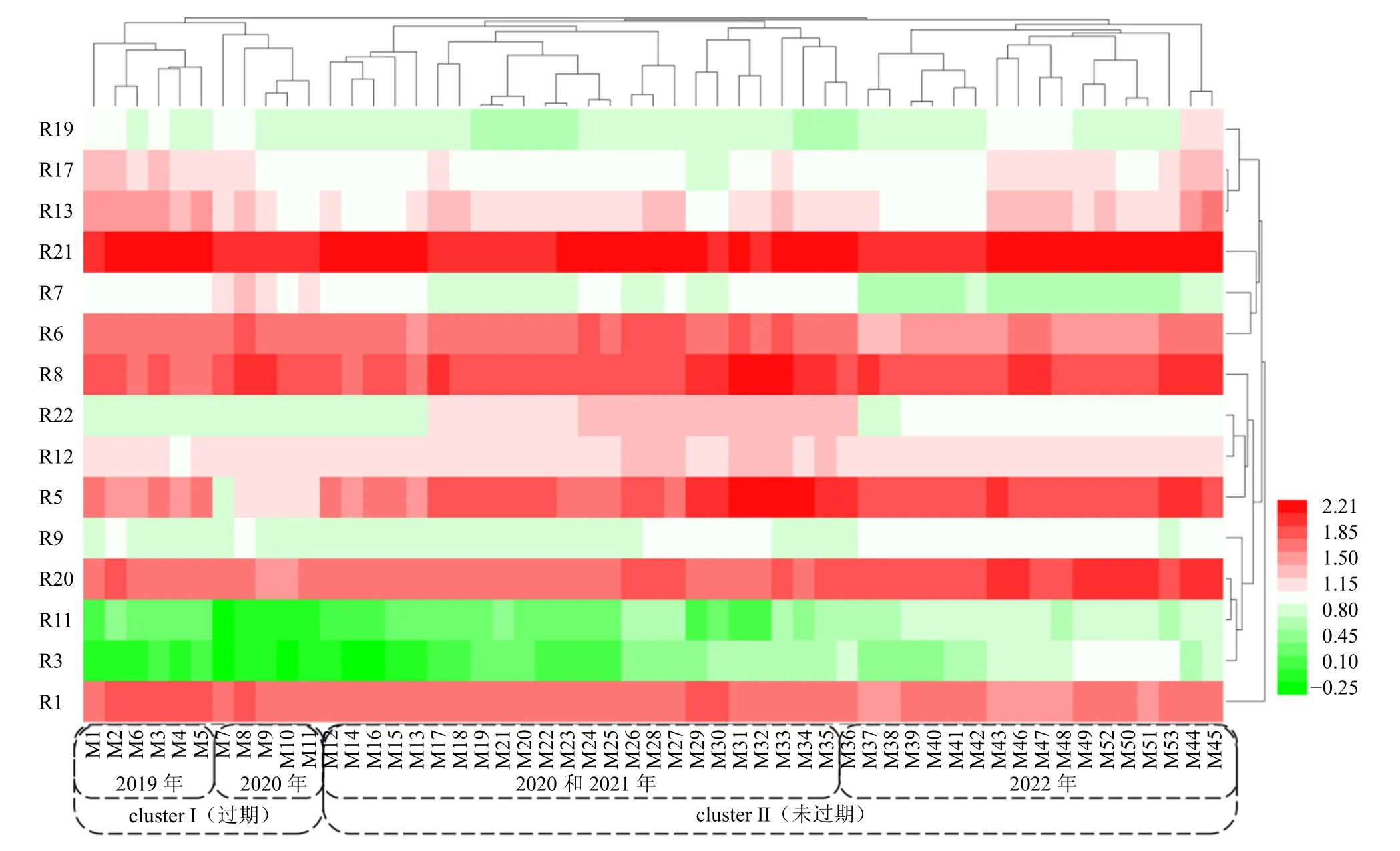
圖6 53 個批次MOL 樣品中15 種成分含量的聚類熱圖Fig. 6 Cluster analysis of contents of 15 compounds in 53 batches of MOL samples
2.6.2 主成分分析(principal component analysis,PCA) 將不同批次樣品各化合物峰面積導(dǎo)入SIMCA 14.1 軟件,進行PCA,見圖7-A 和8-A。玄參藥材樣品大致分為2 類,華北地區(qū)(河北省)玄參藥材樣品單獨為一類,能很好地與其他地區(qū)分開,這與聚類結(jié)果一致,但是其余地區(qū)玄參藥材樣品區(qū)分不明顯,可能是因為第1 和第2 主成分并未包含全部的變量信息。MOL 被分為4 類:2019 年、2020年、2021 年與2022 年的樣品分別聚成一類,說明不同年份的MOL 中各成分存在一定差異,與聚類分析結(jié)果基本一致。
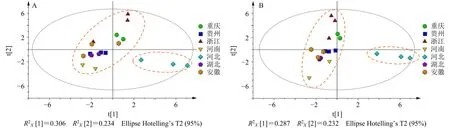
圖7 玄參藥材樣品的PCA 得分散點圖 (A) 和OPLS-DA 得分散點圖 (B)Fig. 7 PCA scatter plot (A), and OPLS-DA scatter plot (B) of Scrophulariae Radix samples
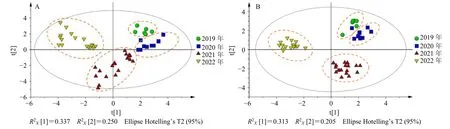
圖8 MOL 樣品的PCA 得分散點圖 (A) 和OPLS-DA 得分散點圖 (B)Fig. 8 PCA scatter plot (A) and OPLS-DA scatter plot (B) of MOL samples
2.6.3 正交偏最小二乘-判別分析(orthogonal partial least squares method-discriminant analysis,OPLS-DA)為更好地區(qū)分不同批次玄參藥材和MOL 樣品組間差異,尋找組間差異性化學成分,進一步對不同批次樣品進行OPLS-DA,見圖7-B 和8-B。玄參藥材樣品和MOL 樣品的分類結(jié)果與PCA 結(jié)果一致,即華北地區(qū)(河北省)的玄參藥材樣品明顯區(qū)分于其他地區(qū);不同年份的MOL 樣品中各成分含量存在一定差異,但同一年份的MOL 樣品中各成分含量相近。
為進一步篩選出不同年份MOL 樣品間差異性化學成分,以變量重要性投影值(variable importance projection,VIP>1)對各成分進行篩選。結(jié)果如圖9 所示,共得到7 個差異性化合物,分別為異毛蕊花糖苷(R13)、對甲氧基肉桂酸(R22)、腺苷(R1)、1-(3-hydroxy-4-methoxyphenyl)ethyl-3-α-Lrhamnopyranosyl-6-caffeoyl-β-D-glucopyranoside(R17)、對羥基苯甲酸(R7)、阿魏酸(R12)和原兒茶酸(R6)。然而,在玄參藥材樣品的OPLS-DA模型中,除河北省(S11~S13)能與其他產(chǎn)地明顯區(qū)分外,其余產(chǎn)地的分類效果并不明顯,所以未進一步采用VIP 值篩選其差異性成分。
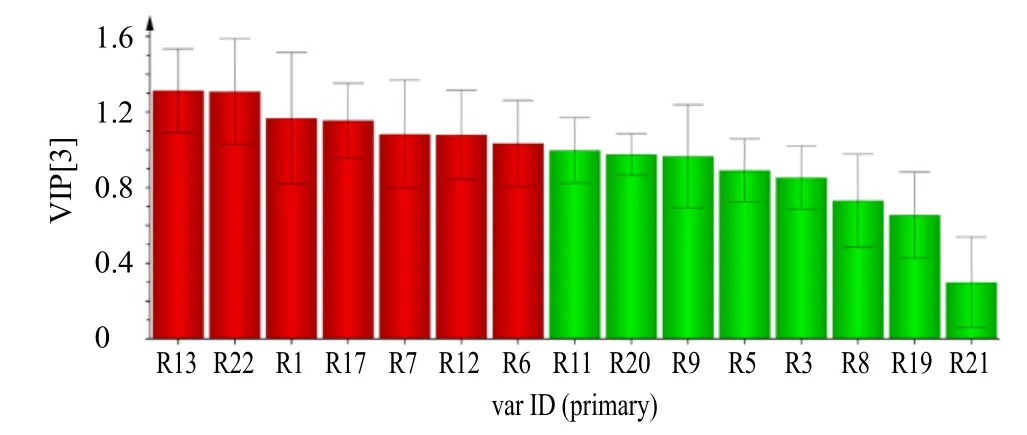
圖9 MOL 樣品中15 種成分含量的VIP 值Fig. 9 VIP values of contents of 15 compounds in MOL samples
3 討論
3.1 指標性成分的選擇
玄參主要以環(huán)烯醚萜苷、苯丙素苷和有機酸類為其主要成分[16]。現(xiàn)代藥理學研究表明,環(huán)烯醚萜苷、苯丙素苷和有機酸具有抗心肌細胞損傷、保護腦缺血損傷和抗動脈粥樣硬化等藥理活性[4,16-19]。環(huán)烯醚萜苷、苯丙素苷和核苷類成分是玄參滋陰的物質(zhì)基礎(chǔ),具有免疫調(diào)節(jié)的活性[6,11,20],其中環(huán)烯醚萜苷和苯丙素苷可通過調(diào)節(jié)陰虛小鼠的能量代謝、免疫力和氧化應(yīng)激而發(fā)揮滋陰的功效。基于此,本研究選擇環(huán)烯醚萜苷、苯丙素苷、有機酸和核苷4 類有效成分作為含量測定的指標性成分。
3.2 玄參藥材供試品溶液提取條件的優(yōu)化
為提高各待測成分在定量分析中的準確度,本研究采用單因素實驗考察提取溶劑(水、甲醇、乙醇)、甲醇體積分數(shù)(30%、50%、70%、100%)、液料比(10∶1、20∶1、40∶1、60∶1、80∶1)、超聲時間(20、30、40、50、60 min)、超聲功率(80、120、160、200 W)及超聲溫度(25、40、55、70 ℃)對玄參中環(huán)烯醚萜苷、苯丙素苷、有機酸和核苷類成分提取率的影響。
環(huán)烯醚萜苷類成分的含量為R4、R5、R16、R20與R23~R25 之和,苯丙素苷類成分的含量為R10、R11、R13~R15 與R17~R19 之和,有機酸類成分的含量為R6~R9、R12 與R21、R22 之和,核苷類化合物的含量為R1~R3 之和。結(jié)果表明,甲醇體積分數(shù)和超聲條件對各類成分的提取率影響不明顯,而料液比則呈現(xiàn)先上升后趨于平緩的趨勢且在60∶1 時各類成分的提取率最大。綜合考慮提取率和時間,本實驗選用70%甲醇,液料比60∶1,超聲處理40 min(200 W、40 ℃)作為玄參樣品的提取條件。
3.3 含量測定結(jié)果的分析
玄參藥材樣品的化學計量學分析結(jié)果表明,CA可很好地區(qū)分4 個地區(qū)玄參藥材樣品,PCA 和OPLS-DA 雖然沒有CA 分類效果好,但也可明顯區(qū)分華北地區(qū)(河北省)和其他地區(qū)的玄參藥材樣品。說明玄參藥材的質(zhì)量受生長環(huán)境影響是客觀存在的,且以華北地區(qū)(河北省)的玄參藥材質(zhì)量最優(yōu)。此外,3 種化學計量學模型表明,不同年份的MOL樣品中各成分含量存在一定差異,但同一年份MOL樣品中各成分含量相近,可能是當年使用的藥材質(zhì)量相對穩(wěn)定。該方法也可為MOL 的質(zhì)量控制提供參考。
此外,本實驗是采用質(zhì)譜技術(shù)對玄參藥材和MOL 樣品中的多類型成分進行定量分析,雖然可為玄參藥材和MOL 的質(zhì)量控制提供科學依據(jù),但使用的儀器昂貴且部分定量的化合物是經(jīng)本課題組分離純化得到。因此,從研究手段和定量成分方面,該方法的普適性還有待進一步加強。但本研究在建立OPLS-DA 模型后,以VIP 值篩選得到的不同年份MOL 的差異性化合物,可作為MOL 的質(zhì)量控制標志物用于其質(zhì)量控制。目前,企業(yè)僅通過HPLC法測定肉桂酸含量評價MOL 的質(zhì)量,檢測的成分單一,今后可考慮加入本實驗中篩選得到的質(zhì)量控制標志物,建立更全面的多成分定量的HPLC 法用于提高MOL 的整體質(zhì)量。
3.4 結(jié)論
本研究基于UPLC-QqQ-MS/MS 技術(shù),首次建立了玄參藥材中多類型成分的MRM 分析方法,并將此方法首次應(yīng)用于MOL 中成分分析。通過化學計量學分析不同產(chǎn)地玄參藥材和不同批次MOL 樣品,為MOL 組方玄參藥材優(yōu)選和MOL 質(zhì)量控制提供參考。
利益沖突所有作者均聲明不存在利益沖突
猜你喜歡
中學生數(shù)理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數(shù)理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數(shù)理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數(shù)理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(shè)(2018年6期)2018-04-22 03:16:54
產(chǎn)品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數(shù)理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
