聯合TCGA和GEO數據庫構建由circRNA介導的非小細胞肺癌特異性競爭性內源性RNA網絡
2023-06-07 05:41:14劉金嬋許德華李讓唐田書陳銘陳曉琳饒紹奇
河北醫藥 2023年7期
劉金嬋 許德華 李讓 唐田書 陳銘 陳曉琳 饒紹奇
在2020年全球癌癥統計中,肺癌是第二常見,也是死亡率最高的癌癥,對人類健康構成了嚴重的威脅。肺癌分為小細胞肺癌(small cell lung cancer,SCLC,15%)和非小細胞肺癌(non-small cell lung cancer,NSCLC,85%)[1];NSCLC早期患者以手術治療為主,Ⅲ期是同步放化療后免疫治療,但患者存活率均較低[2]。因此,找到與NSCLC發生發展相關的分子機制,發現有效的早期診斷生物標志物以及臨床治療的潛在靶點,改善患者的生存和預后,極為重要。circRNA存在共價閉合的單鏈環構象,不易受到破壞,并且存在一個或多個miRNA反應元件(miRNA response element,MRE)[3],可基于競爭性內源性RNA(competing endogenous RNA,ceRNA)機制參與癌癥發展過程中,如circRNAs可作為ceRNA,與miRNA中的MRE結合,減少miRNA與其靶mRNA結合,從而調節miRNA對靶mRNA的抑制作用。先前研究認為circRNA與各種疾病的發展過程都具有顯著地相關性,尤其是關于癌癥的進展過程[4],同時,circRNA在癌癥細胞和各種體液[5]中特異表達,反映出circRNA作為NSCLC診斷生物標志物或治療靶點的極大潛力。在這項研究中,從TCGA數據庫和GEO數據庫獲取的芯片數據,得到NSCLC中差異表達的circRNA、miRNA 和 mRNA并通過這3種RNA來進行ceRNA網絡的構建并識別出網絡中的核心circRNA,并從中提取了NSCLC特異性ceRNA核心子網絡;最后把 ceRNA 機制作為探索非NSCLC發展過程的出發點,來進一步說明核心 circRNA在其中是如何通過調控基因的表達從而作用于癌癥的發展機制,為開展NSCLC的致病機制、診斷生物標志物與治療潛在靶點等研究提供參考。
1 資料與方法
1.1 一般資料 本研究使用的3個circRNA芯片數據GSE101586 (5病例,5對照),GSE101684 (4病例,4對照) 和 GSE112214 (3病例,3對照)均來源于GEO(gene expression omnibus)數據庫。494例NSCLC的miRNA-seq、RNA-seq及臨床信息則來自TCGA(the cancer genome Atlas)數據庫。
1.2 差異表達circRNA (DEcircRNA)的篩選 首先,對GEO數據庫的circRNA數據規范化和以及對數據進行log2的變換,之后采用“limma” R軟件包進行差異表達分析,以|log2(Foldchange)| >1和P-value<0.05為標準確定每個數據集中差異表達的circRNA (DEcircRNA)。然后,使用“Robust Rank Aggreg”R軟件包的穩健的秩集聚法(robust rank aggregation,RRA)根據表達水平和P-value對3個數據集的DEcircRNA進行了整合和排序,確定最優的DEcircRNA。
1.3 差異表達mRNA(DEmiRNA)和miRNA(DEm R
NA)的篩選 首先,對TCGA數據庫NSCLC的RNA-seq和miRNA-seq數據過濾,將表達量較低的基因刪除(過濾標準為:平均表達值<1);隨后,進行差異分析,以FDR校正的P value <0.05和|log2(Foldchange)| >1的標準得到DEmiRNA、DEmRNA的新表達矩陣。
1.4 ceRNA 網絡的構建和核心網絡的提取 首先,從circbase數據庫[6]獲取DEcircRNA的fasta序列和從miRbase數據庫[7]獲取DEmiRNA的fasta序列。隨后,利用miRanda[8]、 RNAhybrid算法[9]預測存在結合靶位的circRNA-miRNA調控關系,兩個算法均預測到的調控對將納入網絡構建。然后,從miRTarbase數據庫[10]中得到DEmiRNA和DEmRNA之間的調控關系。根據對子中相同的 miRNA 進行 ceRNA 網絡的構建,最終得到了多條完整的circRNA-miRNA-mRNA調控軸,根據miRanda算法識別出結合位點數>4的circRNA作為核心節點并提取其調節的子網絡。最后使用 Cytoscape 軟件(版本 3.6.1)可視化。
1.5 功能富集分析和生存分析 首先,利用 “cluster Profile” R包進行GO和KEGG功能通路分析,探討子網絡的生物學功能。然后,將從上述的DEmRNA新表達矩陣和臨床信息整合,再使用 “survival” R包對核心子網中的基因進行Kaplan-Meier分析。
2 結果
2.1 篩選差異表達的 RNA circRNA的差異分析結果顯示,GSE101586、GSE101684、GSE112214芯片分別得到180個、1 341個、587個DEcircRNA。整合3個數據集結果,經RRA算法確定了42個DEcircRNA。RNA-seq得到5 537個DEmRNA。miRNA-seq得到362個DEmiRNA。
2.2 構建非小細胞肺癌特異性circRNA-miRNA-mRNA網絡并提取其核心子網絡 基于miRanda、RNAhybrid算法,本研究得到了642個circRNA-miRNA調控對子;然后,從miRTarbase數據庫獲取了838個miRNA-mRNA調控對子。借助共享的miRNA,構建了包含35個circRNA,77個miRNA,283個mRNA的circRNA-miRNA-mRNA網絡。進一步以結合位點數>4的8個circRNA為核心,并以其調控的分子(miRNA和mRNA)為目標節點,提取了1個包含56個節點(8個circRNA,10個miRNA,38個mRNA)、61條邊的子網絡。見圖1。
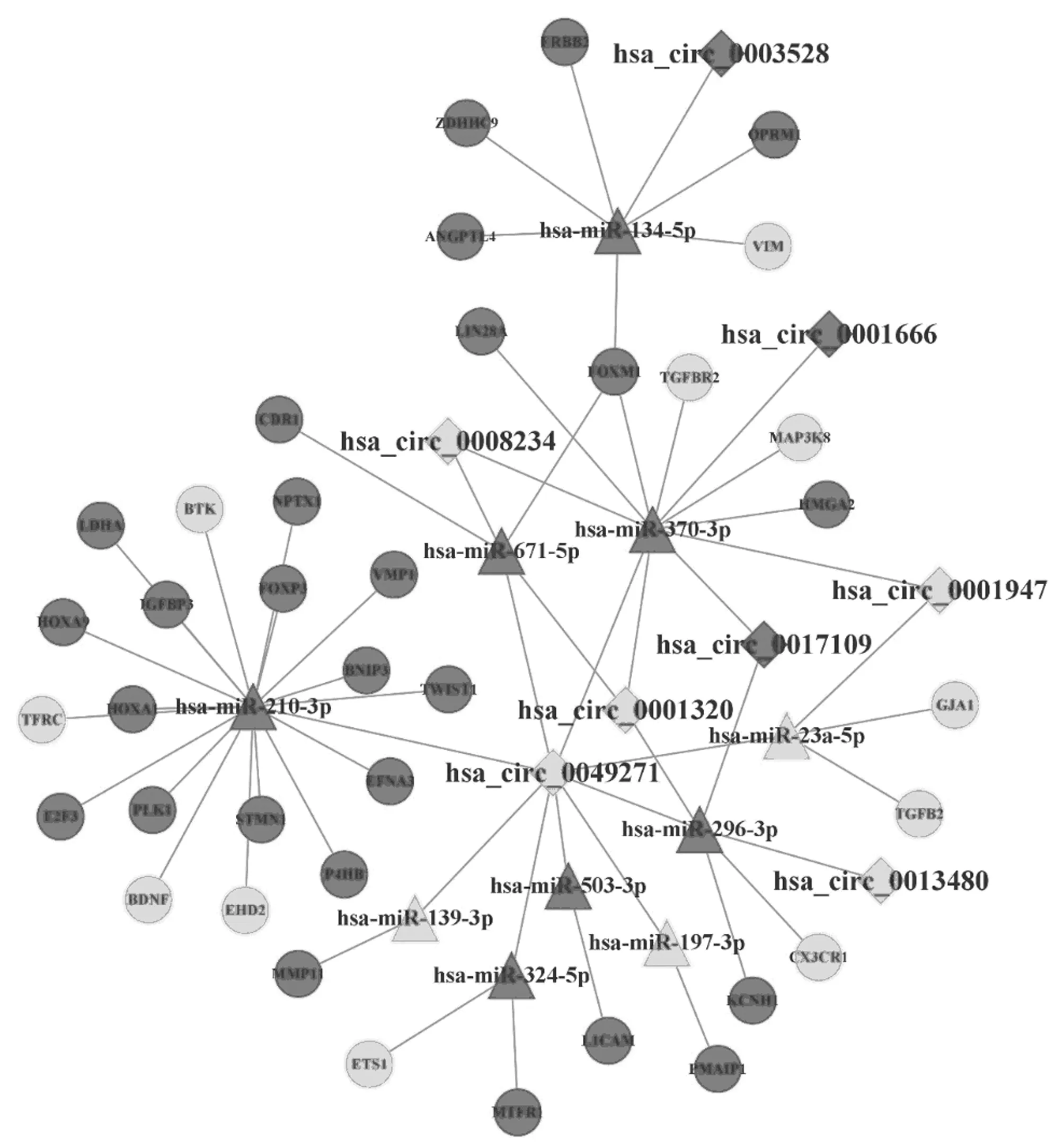
圖1 NSCLC特異性ceRNA核心子網絡;菱形、三角形和圓形分別代表DEcircRNA、 DEmiRNA 和 DEmRNA
2.3 非小細胞肺癌特異性ceRNA核心子網絡的生物
學意義 對GO的富集分析顯示,該ceRNA核心子網絡、顯著富集于由中性粒細胞激活參與的免疫反應、細胞對化學應激的反應、核受體活性,配體激活轉錄因子活性等功能;對KEGG數據庫的富集分析顯示,該ceRNA核心子網絡主要富集于MAPK信號通路、p53信號通路、非小細胞肺癌等通路。以上結果表明NSCLC特異性ceRNA調控網絡涉及多個生物學過程和信號通路。
2.4 非小細胞肺癌特異性ceRNA核心子網絡的的臨床意義 生存分析表明NSCLC特異性ceRNA核心子網絡中有14個基因顯著影響患者的預后情況(P<0.05)。與低表達組比較,ANGPTL4、FOXM1、HMGA2、HOXA1,OPRM1,PMAIP1,LDHA,TWIST1,MTFR1,PLK1基因高表達組的預后較差;而MAP3K8、TGFBR2、BTK、CX3CR1基因高表達組的預后較好(或解釋為保護因子)。見圖2。

圖2 NSCLC特異性ceRNA核心子網絡中14個預后基因標記的生存曲線
3 討論
circRNA作為一種新的診斷標志物和治療分子,已被研究并應用于多種疾病的診斷和治療中。目前多個研究均基于單個cirRNA,而復雜疾病的發展是由于多個生物分子相互作用的復合物的擾動引起的[11]。此外,在ceRNA網絡中,一個circRNA競爭性吸附多個miRNA,一個miRNA也可被多個circRNA吸附,導致下游基因的表達異常。基于以上事實,本研究希望通過構建包含circRNA、miRNA和mRNA的完整ceRNA網絡,系統地闡明NSCLC的病理機制并識別與其預后相關的分子標記。然而在NSCLC的以往研究中,較少有針對circRNA介導的ceRNA調控網絡的全基因組研究。本研究通過充分結合兩個數據庫,構建了由circRNA介導的NSCLC特異性ceRNA調控網絡,從中提取出連通度較高的節點以及由節點所介導的核心子網絡。最后,通過富集分析和生存分析,闡明了NSCLC特異性ceRNA核心子網絡的生物學意義和臨床意義。
ceRNAs和miRNAs共享MRE的數量被證明是ceRNA交叉調節的關鍵決定因素[12],具有異常高密度結合位點的circRNA可能會在疾病網絡中發揮著關鍵作用。本研究利用miRanda算法挖掘到8個核心的circRNA,其中hsa_circ_0001666/0008234/0001947均已被發現與肺癌相關。hsa_circ_0001666高表達與NSCLC 患者淋巴結轉移和病理分期呈正相關[13]。而hsa_circ_0001947則在NSCLC組織和細胞中低表達,敲低后增強了細胞活力和增殖能力,誘導細胞生長時停留在S期,最終導致增殖異常[14]。
8個核心circRNA調控的10個miRNA中,hsa-miR-671-5p、hsa-miR-503-3p、hsa-miR-210-3p、hsa-miR-296-3p、has-miR-197已被報道與NSCLC密切相關,如miR-671-3p在NSCLC組織和細胞系中表達上調[15],通過調節FOXP2促進癌細胞增殖、凋亡和遷移。hsa-miR-503-3p在NSCLC中表達上調,并發現其在癌癥原發性耐藥中發揮重要作用[16]。其余5個miRNA雖暫時未見有報道與NSCLC相關,但均被發現與一種或多種癌癥相關。上述說明本文識別的與circRNA有調控關系的10個miRNA在NSCLC或其他癌癥中發揮重要作用,其生物學意義是明確。
本研究通過對特異性ceRNA核心子網絡進行功能通路富集分析發現,子網絡主要參與到中性粒細胞激活參與免疫反應、配體激活的轉錄因子活性、MAPK信號通路、p53信號通路、非小細胞肺癌等。中性粒細胞是免疫細胞家族的關鍵成員,激活后表達大量膜受體,識別和消除感染因子[17]。KEGG富集結果中的大部分通路已有文獻報道與NSCLC發生機制密切相關,如經典的p53信號通路[18]作為一條抑癌通路廣泛參與到NSCLC中。另外,子網還直接富集到NSCLC的通路上。以上信息提示,本研究所篩選的核心cirRNA介導的ceRNA網絡密切涉及了NSCLC整個發展過程。
生存分析表明NSCLC特異性核心子網絡中有14個基因顯著影響患者的預后情況,而網絡中的8個核心circRNA很可能通過ceRNA調控網來調節這些基因,影響其表達,對患者預后產生作用。例如:在hsa_circ_0001320/0001947/0008234-hsa-miR-370-3p-MAP3K8/TGFBR2調控軸中,cirRNA下調、miRNA上調、mRNA下調,3個circRNA的低表達會減少與miR-370-3p的結合,使更多的miR-370-3p與MAP3K8、TGFBR2直接結合,導致MAP3K8、TGFBR2在體內的含量降低。TGFBR2的缺失會增加腫瘤侵襲性并降低肺腺癌的存活率[19]。
而本文生存分析的結果以及相關研究[20]都表明,MAP3K8、TGFBR2基因表達量低的患者,生存率相對較低。因此,推測hsa_circ_0001320/0001947/0008234等circRNA的低表達可能與NSCLC患者的不良預后相關。在hsa_circ_0049271-hsa-miR-210-3p-BTK這一條調控關系上,circ_0049271表達下調、miR-197-3p、BTK表達上調,說明circ_0049271下調可導致BTK的低表達,BTK調節p53的活性以增強細胞凋亡、衰老反應以及腫瘤抑制反應[21]。生存分析同時也顯示BTK低表達組患者預后較差,提示hsa_circ_0049271低表達與NSCLC患者不良預后相關。總之,本文通過構建NSCLC特異性ceRNA核心子網絡識別出來的8個circRNA可能通過與一個或多個miRNA結合,以此來調節與預后顯著相關基因的表達,進而影響NSCLC患者總體預后情況。
然而,我們也注意到本研究的不足之處。(1)用于篩選差異表達circRNA的三套數據樣本量偏小,可能會缺少足夠的統計功效識別微小或中度效應的circRNA。(2)部分circRNA、miRNA和mRNA之間的調控關系是通過計算機算法識別的,缺少實驗室的證據。(3)研究所選的數據針對西方群體,因此本次研究是否能推廣和應用到中國人群中,仍然需要進行探討。本研究通過整合多組學的數據,系統性分析了circRNA、miRNA和mRNA之間的調控關系,并構建NSCLC特異性ceRNA核心子網絡。進一步功能富集分析和生存分析表明NSCLC特異性ceRNA核心子網絡具備清晰的生物學意義和臨床意義,在8個circRNA介導的基因中,有14個基因顯著影響患者的預后情況。
猜你喜歡
保健醫苑(2023年2期)2023-03-15 09:03:04
中國臨床醫學影像雜志(2022年2期)2022-05-25 13:24:34
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
癌變·畸變·突變(2016年3期)2016-02-27 06:15:34
財經(2016年6期)2016-02-24 07:41:51
醫學研究雜志(2015年12期)2015-06-10 06:57:46
鄭州大學學報(醫學版)(2015年1期)2015-02-27 14:50:26
