AP在氟化石墨烯表面分解機理的密度泛函理論研究
2023-03-11 09:52:28楊秀榮張梓涵趙子航李嘉辰馬海霞
火炸藥學報 2023年2期
關鍵詞:結構
張 馳,楊秀榮,張梓涵,趙子航,李嘉辰,馬海霞
(西北大學 化工學院&西安特種能源材料重點實驗室,陜西 西安 710069)
引 言
隨著對導彈和火箭發動機的要求不斷提高,除了改善其發動機的機械結構,提高導彈和火箭發動機的主要能量材料——復合固體推進劑的性能同樣是一種可行且高效的手段。高氯酸銨(AP)作為廣泛使用于復合固體推進劑配方中的一種氧化劑,在推進劑中的用量所占的質量分數為60%~90 %,其燃燒性能對復合固體推進劑的燃燒行為有重要影響[1-3]。例如,推進劑的燃燒穩定性、推力等性能會受到AP的分解活化能和高溫分解溫度等性質的顯著影響[4-7]。大量研究表明添加劑的使用能夠明顯改善AP的燃燒性能[8],激發了研究者對AP分解反應機理的探索[9-17]。研究表明,在AP熱分解過程中,由于分解生成的NH3分子會吸附在AP晶體表面,隨著NH3吸附量的增加,會阻止AP晶體持續分解生成NH3和HClO4分子,從而阻止AP的完全分解。隨著溫度的升高,吸附在AP表面的NH3分子逐漸脫附,AP繼續分解,直到反應完全。因此導致純AP的熱分解被分割為低溫分解階段(LTD)和高溫分解階段(HTD)[18-21]。這種熱行為會導致AP基固體推進劑的點火延遲時間延長,燃燒速度較低[22-23]。因此選擇合適的AP燃燒催化劑對消除阻滯期以及提高固體推進劑的性能有重要影響。采用理論計算方法研究AP在催化劑表面的反應,了解AP在催化劑表面分解的具體反應機理,可以避免實驗后催化劑性能不理想而造成的物質浪費,并且能夠對后續可能的實驗研究起到啟發和指導作用。
氟化石墨烯(FG)是石墨烯的部分或全部碳原子通過與氟原子形成共價鍵后生成的二維層狀石墨烯衍生物。FG具有高耐熱性、高導熱性、自由基特性等[24-27]。因此FG作為一種石墨烯功能化改性材料逐漸受到廣泛地關注以及研究,并且其在電子器件(超級電容和傳感器等)、化學電源、生物材料以及催化材料等諸多領域都展現出了良好的應用前景[28-30]。含氟炭材料如聚四氟乙烯和氟化石墨等由于C—F共價鍵具有高強度和高能量,可以用于提高固體推進劑燃燒效率[31-32]。而FG不僅具有可調控密度的C—F共價鍵,同時具備二維片層結構,有望作為包覆劑用于含能材料、固體推進劑等領域[33-35]。
本研究對AP與FG間的相互作用進行了分子層面的探討,分析了將FG用于AP燃燒催化的可行性。采用密度泛函理論方法對AP在FG表面的分解機理進行了研究。分別計算了AP的初始分解產物HClO4(PA)和NH3分子在FG表面的分解歷程。對于HClO4分子,基于文獻提出了兩條不同的分解歷程以及可能發生的基元反應,根據過渡態理論對這些基元反應進行了研究。通過對計算得到的所有基元反應的活化能以及反應熱的分析,建立了HClO4分子在FG表面分解的反應網絡并探討了HClO4分解的具體分解路徑。采用同樣的方法研究了NH3分子的分解行為。最后,對HClO4和NH3分子間可能的相互影響進行了討論,以推測AP整體在FG表面的分解情況。通過對AP在FG表面分解機理的分析,有助于了解催化劑促進AP分解的具體原因以及作用方式,進而為運用理論計算方法篩選出促進AP分解催化劑提供理論依據。
1 計算方法及模型
密度泛函理論經過Thomas-Fermi模型的提出、Hohenberg-Kohn理論的證明,最終通過Kohn-Sham方程的建立逐漸發展并最終進行實際的應用。DFT方法的總能量的表達式如式(1)所示:
E(ρ)=ET(ρ)+EV(ρ)+EJ(ρ)+EXC(ρ)
(1)
式中:ET、EJ、EV和EXC分別表示電子動能、庫倫作用能、電子與原子核間吸引勢能以及交換-相關能。伴隨著泛函梯度的引入,交換-相關能變得更加精確而得到廣泛的使用。
本研究所有理論計算均使用基于密度泛函理論開發的Materials Studio軟件(Version 8.0)中的Dmol3模塊完成[36]。采用了廣義梯度近似(GGA)[37]中的Perdew-Burke-Ernzerhof (PBE)[38]交換關聯勢計算了電子交換關聯能。采用迭代子空間普洛伊的直接反演技術(DIIS)加速系統的SCF迭代速度。根據系統的對稱性,使用Monkhorst-Pack網格計算方法對布里淵區進行采樣,在進行結構優化時將網格k點設置為2×2×1,而在進行性質計算時則將網格k點設置為3×3×1。采用DFT+D中的Grimme van der Waals方法對AP分子及其分解產物與氟化石墨烯表面間的分子間弱相互作用進行校正[39]。在搜索能量最低的幾何結構的過程中,能量、力、位移和自洽場(SCF)的收斂標準分別設定為1.0×10-5Ha(1Ha=27.21eV),0.002Ha/?,0.005?和1.0×10-6Ha。根據Methfessel-Paxton方案確定了電子占用率,為0.001Ha。在研究反應機理的過程中,采用完全線性同步聯合二次同步(complete Linear Synchronous Transit/Quadratic Synchronous Transit,complete LST/QST)方法對各基元反應的過渡態進行搜索,隨后通過頻率計算及NEB方法確定所得到的過渡態結構是屬于最低能量路徑(MEP)上的正確過渡態[40]。
反應活化能Ea以及反應熱Er的定義如式(2)及式(3)所示:
Ea=ETS-EIS
(2)
Er=EFS-EIS
(3)
式中:EIS、ETS和EFS分別為反應初始態(initial state,IS)結構、過渡態(transition state,TS)結構以及最終態(final state,FS)結構的總能量。
吸附質在催化劑表面的吸附能(Ead)根據式(4)進行計算:
Ead=EB/surface-(Esurface+EB)
(4)
式中:EB/surface、Esurface和EB分別為吸附體系、催化劑表面以及獨立的吸附質的總能量。當吸附能為負值時,表示吸附質在吸附劑上的吸附是穩定的,吸附過程為放熱反應。
根據文獻報道[41],在F原子含量相同的情況下,雙面吸附的FG的結構比單面吸附更穩定,同時當FG的原子百分比濃度為25%時(原子比率C∶F為4∶1),FG能夠穩定存在。因此本研究構建了4×4的FG超晶胞(包含32個C原子和8個F原子),為防止層間相互作用,沿z軸方向設立了15?的真空層,氟化石墨烯表面結構如圖1所示。
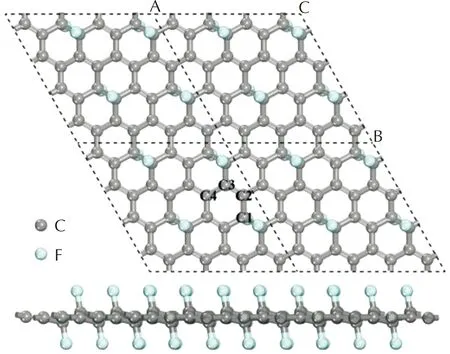
圖1 氟化石墨烯(FG,CF0.25)晶胞結構的頂視圖和側視圖Fig.1 The supercell structure of single-layer FG
由于在構建FG時,并未對晶胞進行固定,幾何優化后的FG晶胞相比于潔凈的石墨烯晶胞有輕微的晶胞參數變化,晶胞參數列于表1。

表1 單層氟化石墨烯以及石墨烯超胞晶胞參數Table 1 The geometric parameters of single-layer FG and pristine graphene supercell
由于F原子含量較低,F原子修飾后的石墨烯晶胞僅有輕微擴大。由于F原子在石墨烯表面的吸附,與F原子直接相連的C原子沿z軸方向向F原子的方向輕微移動,這導致石墨烯表面上C—C鍵的鍵長發生顯著變化,在潔凈的石墨烯超晶胞中,C—C鍵的鍵長平均為1.42?,而在FG超晶胞中,C—C鍵的鍵長處于1.374~1.528?范圍內。根據超晶胞以及FG的對稱性,FG表面共有4種不同化學環境的C原子,這4種C原子分別以羅馬數字標記于圖1中。
2 結果與討論
2.1 HClO4及其分解產物在FG表面上的吸附
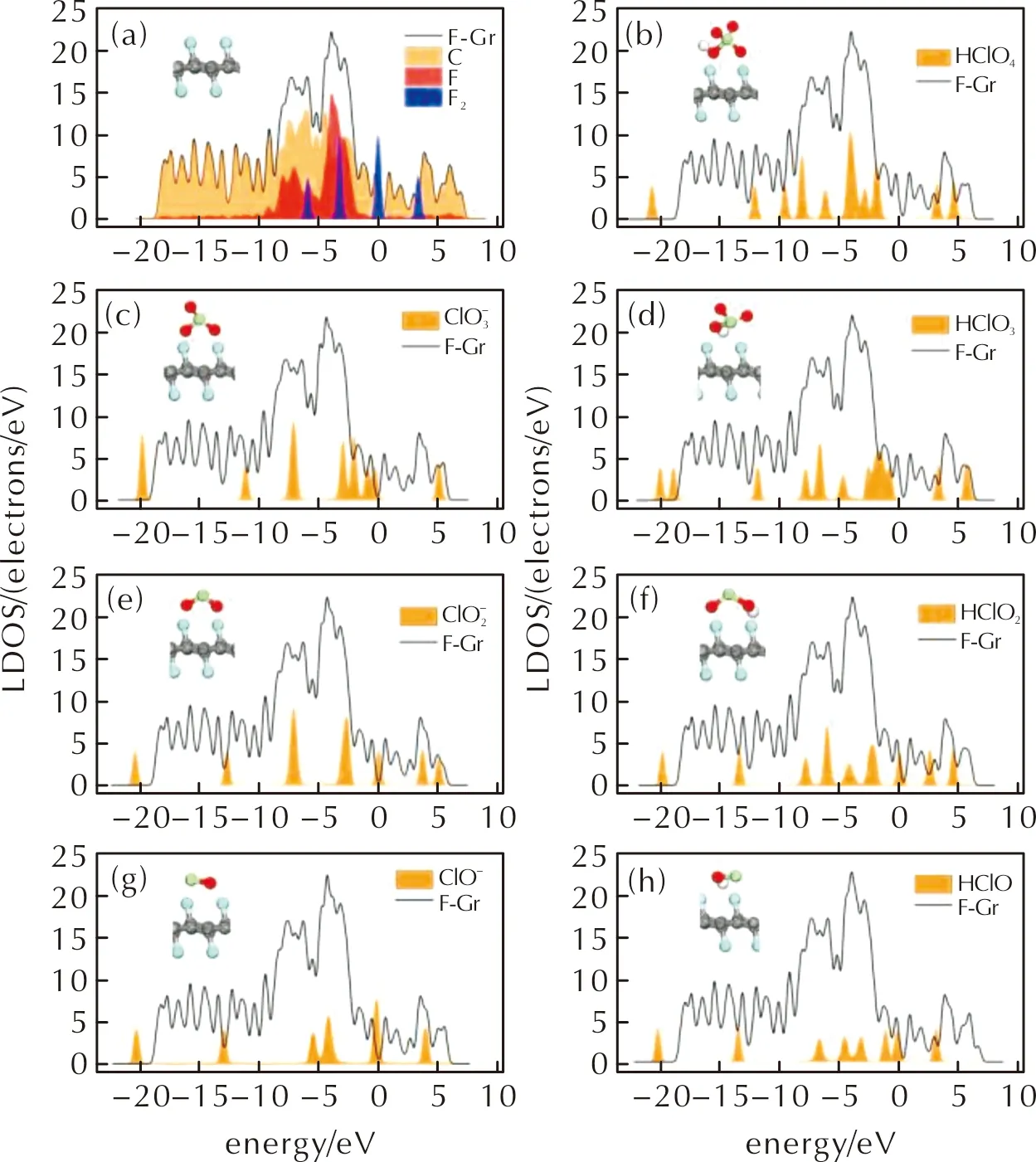
圖2 (a)潔凈的FG表面和(b~h)HClO4及其分解產物在FG表面上吸附結構的側視圖及其局域態密度(LDOS)圖Fig.2 (a) Adsorption structures and local density of states for clean FG surface and (b—h) HClO4 as well as its decomposition products on FG surface
由圖2(a)可知,隨著F原子在石墨烯表面上的吸附,F原子的態密度峰發生雜化,與石墨烯上的碳原子DOS峰發生重疊,F原子的DOS峰廣泛分布于C原子的分布能級-20~7eV范圍內,并主要與能量在-9~-2eV能級處的C原子DOS峰重疊。F原子DOS峰與C原子DOS峰之間的全面重合,表明了F原子在石墨烯表面的化學吸附。由圖2(b)可知,當HClO4吸附在FG表面時,HClO4分子的DOS峰與FG的DOS峰完全沒有重疊,而吸附了HClO4分子的FG的DOS峰的數目與能級基本與潔凈的FG表面的DOS峰一致,表明當HClO4分子吸附在FG表面時,兩者間幾乎沒有發生電子的轉移(計算得到的HClO4分子的mulliken電荷數為0.002e)。HClO4分子在FG表面吸附的幾何結構和電子結構均表明HClO4分子的吸附為物理吸附,因此HClO4分子在FG表面的擴散及移動是相對容易發生的。如圖2(c~h)所示,HClO4分子分解產生的各種中間產物在FG表面的吸附結構的LDOS圖與HClO4分子的LDOS具有類似的情況,表明這些中間產物均為物理吸附。

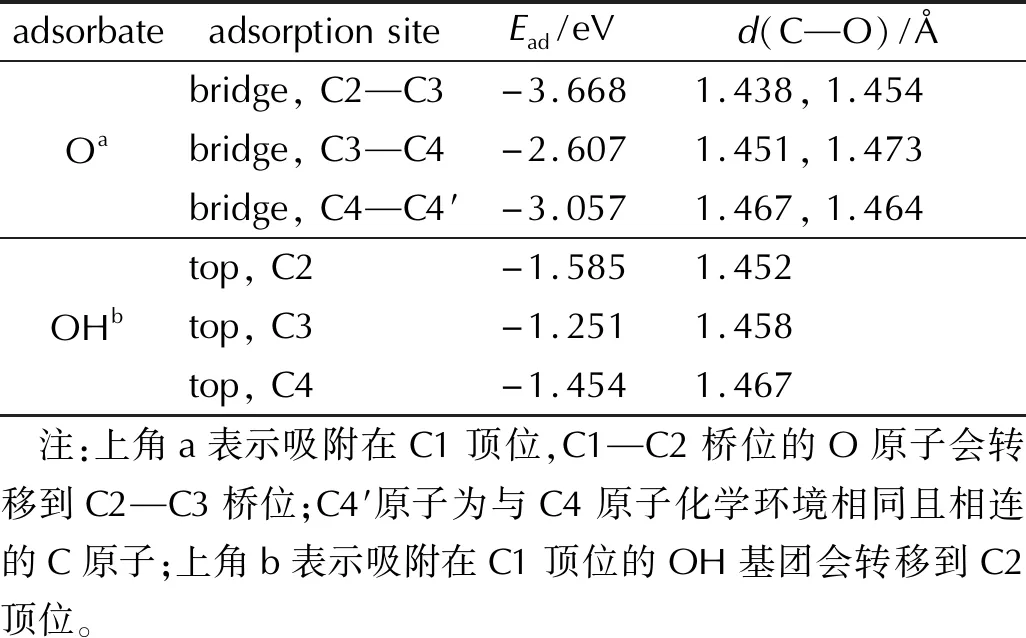
表2 氧原子及羥基基團在FG表面上的各吸附位點的吸附能以及C—O鍵鍵長Table 2 The adsorption energies and C—O bond lengths of oxygen and hydroxyl group on FG surface
當O原子吸附在FG表面時,在C原子頂位吸附的O原子會轉移到C原子橋位與兩個橋頭C原子形成兩個C—O鍵,吸附在不同位置的O原子經過完全優化后得到3種穩定的吸附構型,其中O原子吸附在C2—C3原子橋位時,吸附能的絕對值最大,O原子與FG間的相互作用最強,負值表示吸附后體系能量降低,吸附構型是穩定結構。因此后續計算中,O原子均吸附在C2—C3橋位。對OH基團在FG表面不同位點的吸附構型進行完全優化,部分吸附構型中吸附位點上的OH基團在優化過程中會發生轉移,最終得到3種穩定的吸附結構。當OH基團吸附在C2原子頂位時,吸附能的絕對值最大,吸附結構最穩定。因此在后續計算中,OH基團均吸附在C2原子頂位。
2.2 HClO4分解的酸根路徑
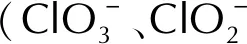
對HClO4分子在潔凈的FG表面沿酸根路徑進行分解的各個基元反應進行過渡態搜索,計算各反應的活化能以及反應熱,確定最低能量路徑(minimum energy path,MEP)。HClO4分子在FG表面的分解主要是分子中的Cl—O鍵的伸長以及斷裂。選擇HClO4分子及其分解的中間產物在FG上的吸附結構作為各基元反應的IS結構,各IS結構中的吸附質分子發生Cl—O鍵斷裂后,解離的O或OH吸附在FG表面上的穩定結構作為FS結構。通過過渡態搜索得到的各個基元反應的過渡態結構僅有一個虛頻,同時過渡態確認的結果與過渡態搜索得到的過渡態結構相一致,表明得到的過渡態結構是能量最低路徑上的正確過渡態。沿酸根路徑上各個基元反應的IS、TS和FS結構如圖3所示,相應的反應能量曲線如圖4所示。
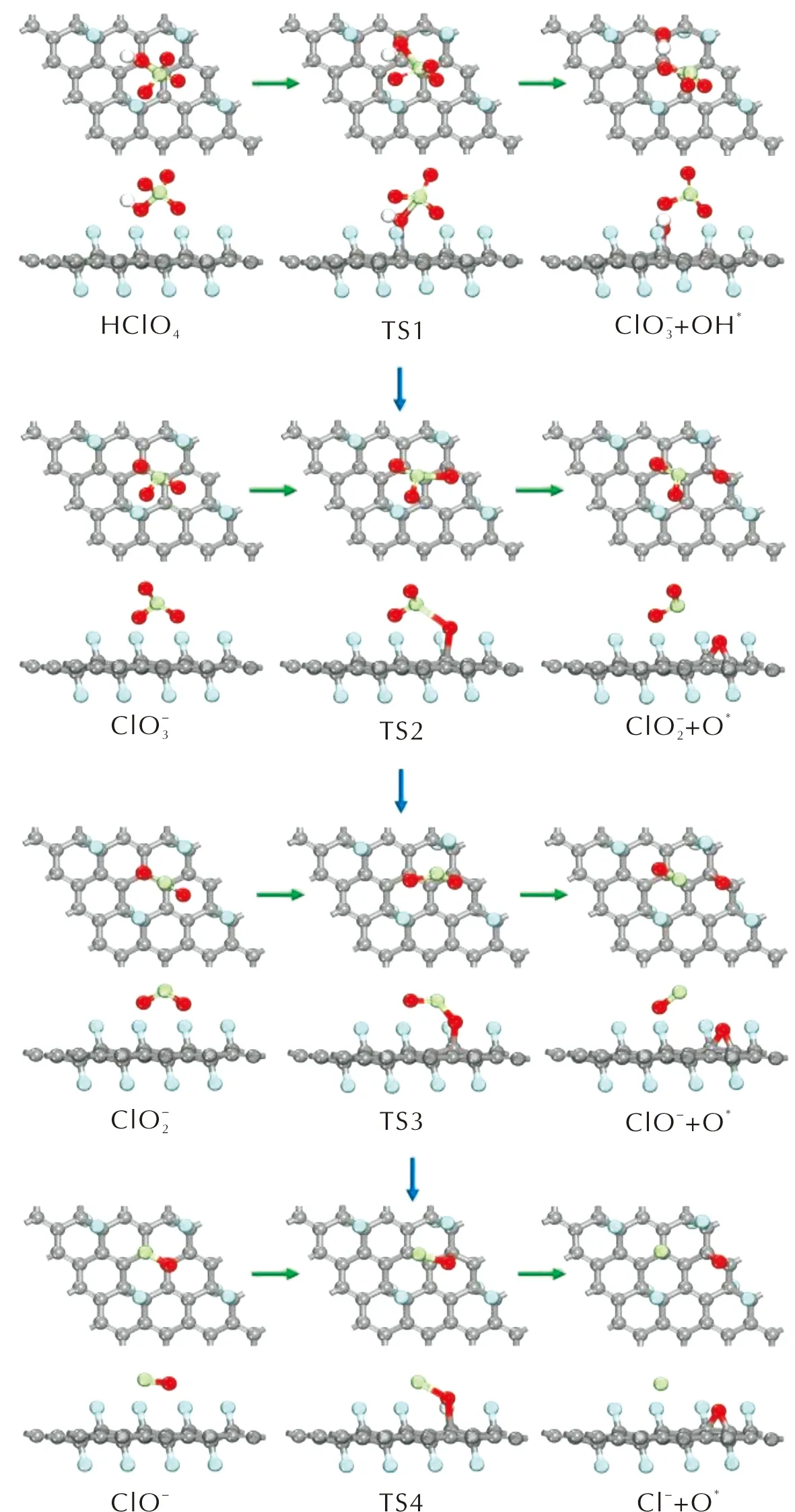
圖3 HClO4在氟化石墨烯上沿酸根路徑反應的各基元反應的結構圖Fig.3 Optimized structures of decomposition reactions of PA and its intermediate products along the pathway that generated oxychloride on FG surface
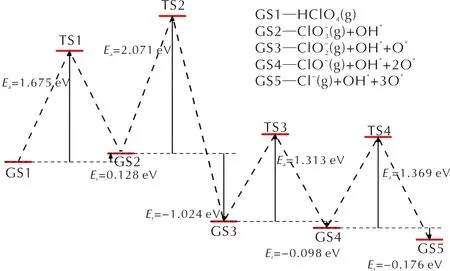
圖4 HClO4在FG上沿酸根路徑的各基元反應的能量曲線Fig.4 Energy profile of decomposition reactions along the chlorate route that generated oxychloride firstly


圖5 HClO4分解產物在氧化FG表面反應的優化結構圖Fig.5 Optimized structures of decomposition reactions of intermediate products of PA along the pathway that generated oxychloride on oxidized FG
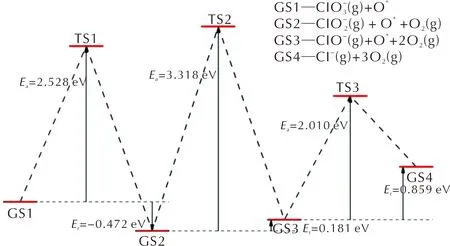
圖6 HClO4分解產物在氧化FG上的基元反應的能量曲線Fig.6 Energy curves of decomposition reactions of intermediate products of PA on oxidized FG
2.3 含氧氯酸路徑
催化劑的使用會改變物質分解的分解路徑,研究發現,部分AP燃燒催化劑的使用會導致AP的分解產物中檢測到HClO2和HClO分子的存在[43-44]。有研究者提出HClO4分子分解時會直接發生Cl—O鍵斷裂生成HClO3分子的反應[45-46]。基于這種情況,HClO4分子除沿著酸根路徑進行分解外,還可能沿下列反應路徑進行分解,該反應路徑記為含氧氯酸路徑:
通過過渡態搜索方法對該分解路徑上的各個基元反應進行了計算,得到的IS、TS和FS結構如圖7所示,HClO4分子沿含氧氯酸路徑分解的能量變化如圖8所示。
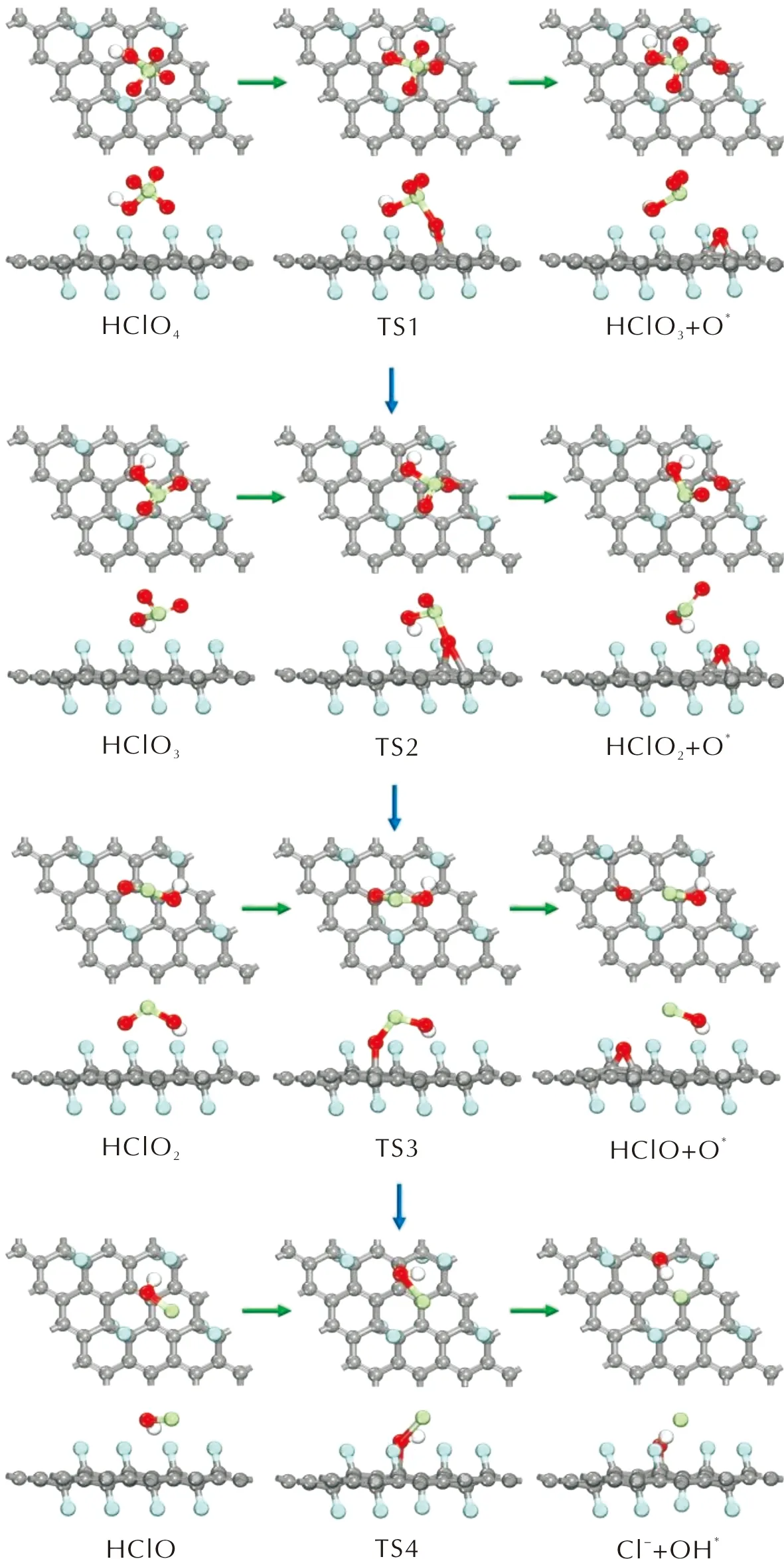
圖7 HClO4分子沿含氧氯酸路徑分解的各基元反應的優化結構圖Fig.7 Optimized structures of decomposition reactions of PA along the oxychloric acid route
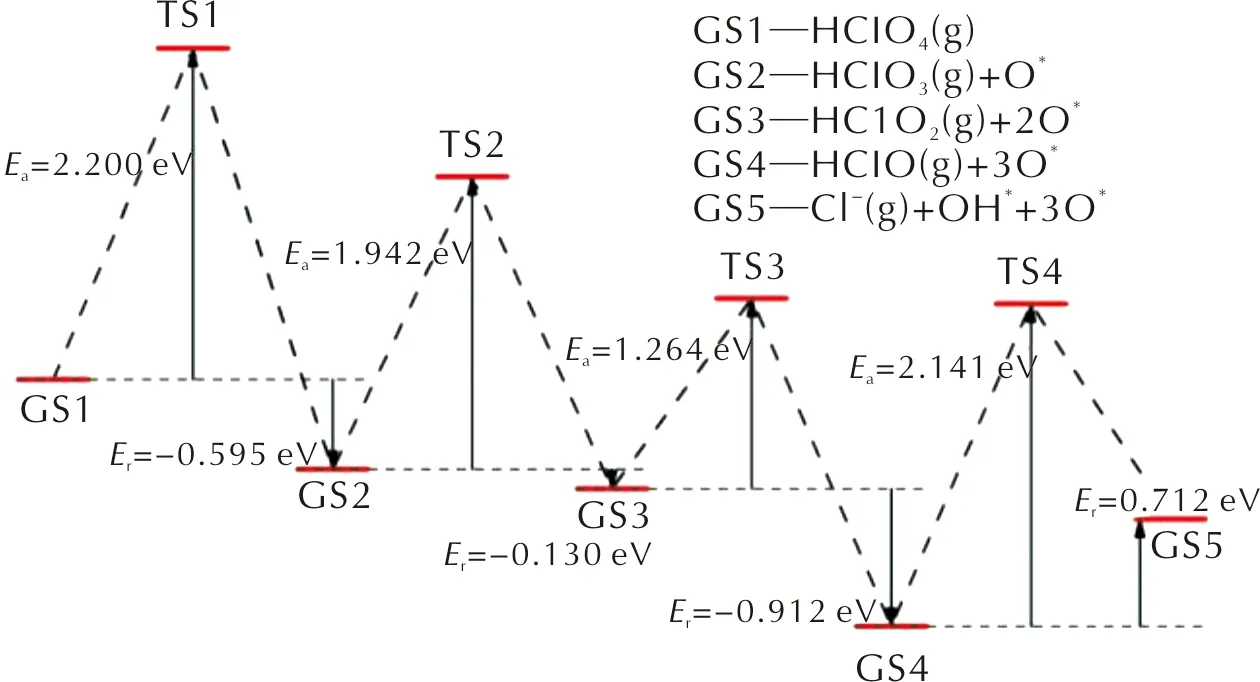
圖8 HClO4分子在FG表面沿含氧氯酸路徑分解的各基元反應的反應勢能曲線Fig.8 Energy curves of decomposition towards elementary reactions of PA on FG surface along the oxychloric acid route
沿含氧氯酸路徑,HClO4分子在FG表面首先發生Cl—O鍵斷裂,解離出一個O原子并生成HClO3分子,解離出的O原子與表面C原子形成C—O鍵。該基元反應為放熱反應,其活化能為2.200eV,反應熱為-0.595eV。根據上述討論,吸附在FG表面的O*原子會抑制Cl—O鍵的斷裂反應。因此,新生成的HClO3分子在FG表面上擴散至未吸附O原子或OH基團的區域繼續進行Cl—O斷裂反應。HClO3分子經過Cl—O斷裂反應生成HClO2分子和吸附在FG表面的O*原子,反應的活化能壘為1.942eV,反應熱為-0.130eV。隨后,生成的HClO2分子同樣經過擴散-分解過程,通過Cl—O鍵的斷裂生成HClO分子和O*原子。HClO2分子發生Cl—O鍵斷裂反應時需要克服1.264eV的活化能壘,當解離出的O*原子與FG表面的C原子形成C—O鍵后,反應體系向外界釋放出0.912eV的能量。最后,HClO分子中羥基的Cl—O鍵斷裂生成Cl-離子和OH基團,OH基團中的O原子與表面C原子形成C—O鍵而吸附在FG表面,Cl-離子與OH*基團形成氫鍵,從而以物理吸附的方式吸附在FG表面。HClO分子的分解反應是一個吸熱反應,反應的活化能和反應熱分別為2.141和0.712eV。
2.4 酸根路徑與含氧氯酸路徑間的轉換
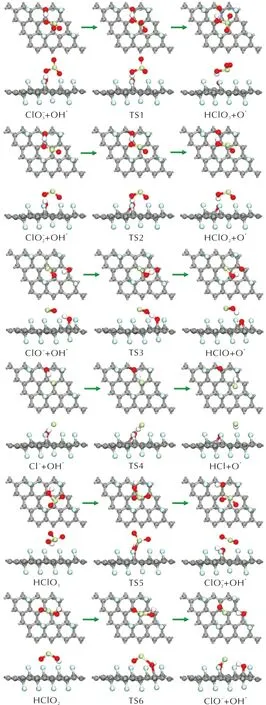
圖9 酸根路徑與含氧氯酸路徑上的中間產物間發生相互轉化的各個基元反應的結構圖Fig.9 Optimized structures of possible elementary reactions for mutual conversion between the intermediate products of acid radical path and oxychloric acid path
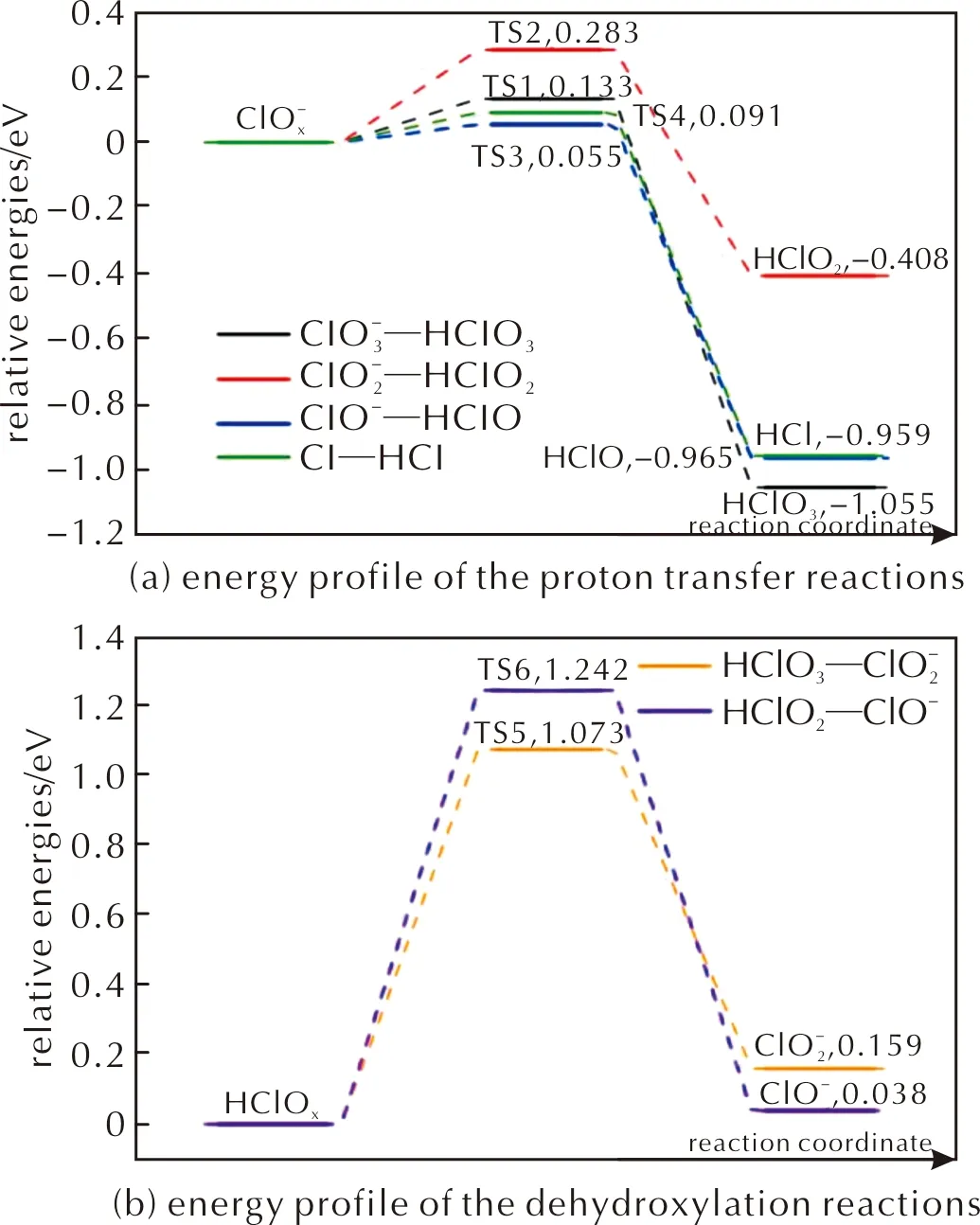
圖10 酸根路徑與含氧氯酸路徑上的中間產物間發生相互轉化的各個基元反應的能量曲線Fig.10 Energy profile of possible elementary reactions of intermediate products of PA on FG expect the chlorate route and oxychloric acid route
2.5 HClO4在FG表面分解反應的機理分析
從上述討論可以發現,當HClO4分子在FG表面的進行分解時,酸根路徑和含氧氯酸路徑上的中間產物在一定條件下均有反應生成另一條分解路徑上的其他中間產物的趨勢。為了明確HClO4分子在FG表面上的具體分解路徑,了解分解反應過程中反應物的消耗、中間體和產物的形成,以及各個中間產物間可能的反應關系,獲得HClO4分子在FG表面上發生分解反應的反應網絡圖。該反應網絡包含了酸根路徑、含氧氯酸路徑以及兩條反應路徑上的中間產物相互轉化的各個基元反應,如圖11所示。

圖11 HClO4在FG表面上分解反應的反應網絡圖Fig.11 The reaction network of the decomposition of PA on FG surface
2.6 NH3在FG表面上的連續脫氫反應
NH3分子作為AP分解的重要初級中間產物,其在FG表面的分解行為是AP分解機理的重要組成部分,同時NH3分子在FG表面的脫氫反應不僅會受到HClO4分子分解的影響,也會影響HClO4分子在FG表面的分解反應。由于HClO4分子在FG表面分解時會有大量O*原子和一定數量的OH*基團吸附在催化劑表面上,因此,不僅研究了NH3分子在潔凈的FG表面的分解行為,同時也研究了其在吸附了O*原子和OH*基團的FG表面的脫氫反應。
首先研究了NH3分子在3種FG表面上的吸附情況,NH3在潔凈的、預吸附了O*原子或OH*基團的FG表面的吸附結構如圖12中的IS結構所示。在潔凈的FG表面上,NH3分子與C4原子形成C—N鍵,吸附能為0.073eV。O*原子的存在會抑制NH3分子在FG表面的化學吸附,此時吸附能為0.114eV。而當OH*基團吸附在FG表面時,則會促進NH3分子在催化劑表面上的吸附,吸附能為-0.013eV。
對已經通過C—N鍵吸附在3種FG表面的NH3分子的脫氫反應進行過渡態搜索,各個基元反應中的IS、TS和FS結構如圖12所示,反應活化能以及反應熱列于表3。
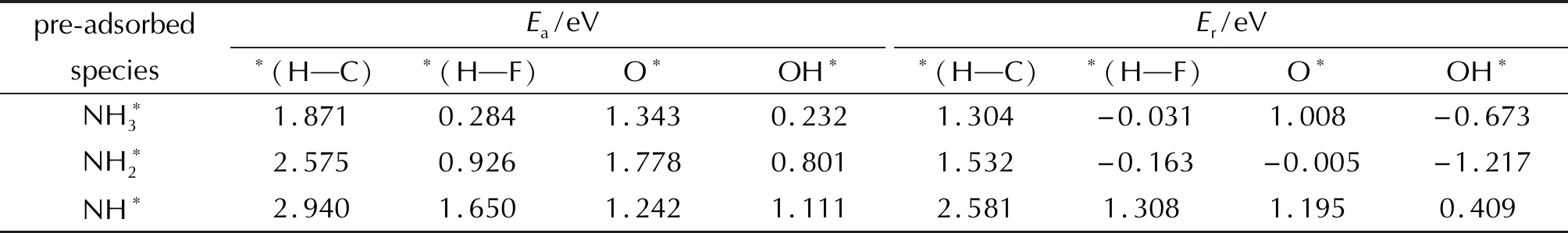
表3 NH3分子在FG表面的脫氫反應的活化能Ea及反應熱ErTable 3 The activation energy (Ea) and reaction heat (Er) of NH3 dehydrogenation on FG surfaces
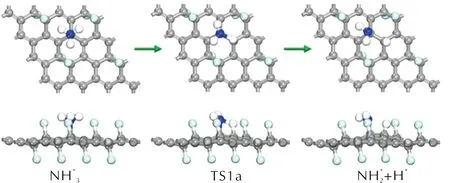
圖分子在FG表面上脫氫反應的幾何結構Fig.12 The optimized structures of dehydrogenation reactions of adsorbed molecule on FG surfaces
NH3分子在潔凈的FG表面發生N—H鍵斷裂,H原子與相鄰的C原子形成C—H鍵,該基元反應是一個吸熱反應,活化能和反應熱分別為1.871和1.304eV。而當O*原子存在時,NH3分子與O*原子反應生成NH2和OH*基團的反應活化能降低為1.343eV,反應仍為吸熱反應,但是體系需要吸收的能量降低為1.008eV。OH*基團的存在會進一步降低NH3分子在FG表面發生脫氫反應的活化能。NH3分子中的一個H原子轉移至OH*基團上形成H2O分子,隨后C—O鍵斷裂,形成的H2O分子從FG表面脫附。該基元反應是一個放熱反應,活化能和反應熱分別為0.232和-0.673eV。O*和OH*的存在均能夠活化NH3分子的N—H鍵,促進NH3的脫氫反應,有助于AP分解過程中產生的游離的NH3分子的分解。

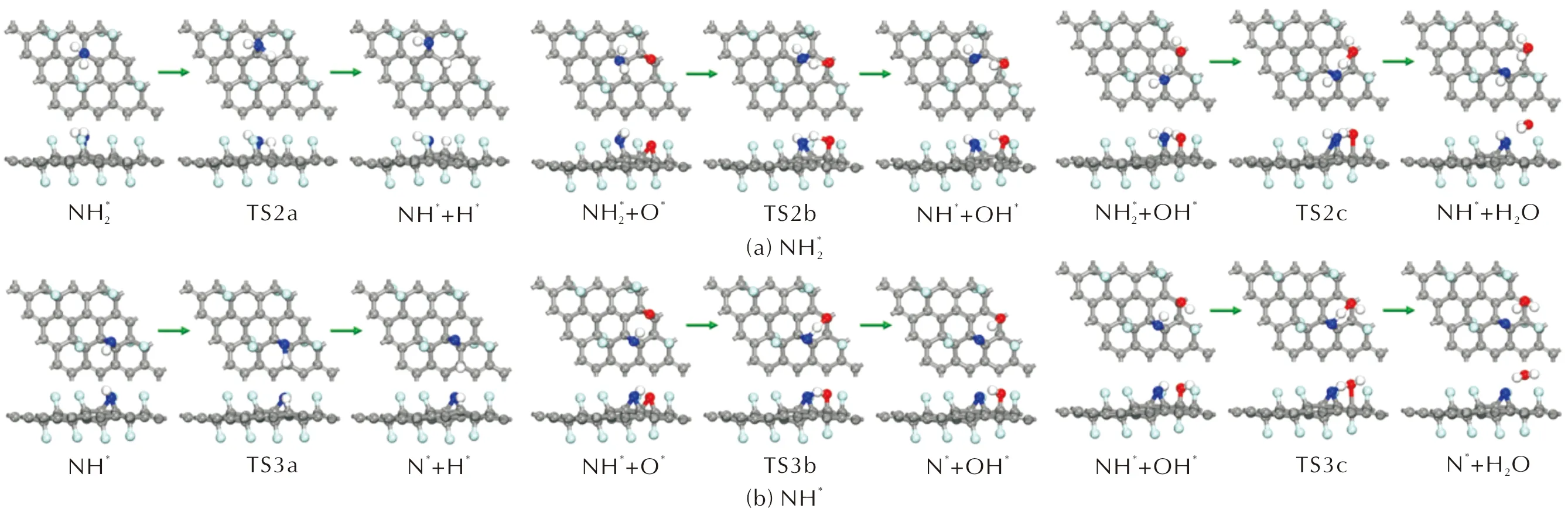
圖和NH*在FG表面上的脫氫反應的幾何結構Fig.13 The optimized structures of dehydrogenation reactions of and NH* on FG surfaces
不論是在潔凈的FG表面還是預吸附了O*原子或OH*基團的FG表面,NH*發生N—H鍵斷裂反應的反應歷程同樣與NH3是相同的。對于NH3、NH2和NH,隨著脫氫反應的逐步進行,N—H鍵斷裂反應的活化能逐漸提高。NH*在潔凈的預吸附OH*的FG表面的脫氫反應活化能分別提高至2.940和1.111eV,反應熱分別為2.581和0.409eV。而NH*與FG表面上吸附的O*原子反應生成N*和OH*的反應的活化能相比于NH3和NH2有所降低為1.242eV,反應熱為1.195eV。
當NH3分子在潔凈的FG表面進行脫氫反應時,解離的H原子除了與表面C原子形成C—H鍵外,還可能與F原子形成F—H鍵,F—H鍵的形成會導致C—F鍵的斷裂,從而形成HF分子。形成的HF分子通過H原子與N*原子間形成的氫鍵以物理吸附的方式吸附于FG表面。對NH3在潔凈的FG表面發生脫氫反應生成NHx和HF的基元反應進行過渡態搜索,反應過程中的優化結構如圖14所示,各反應的活化能及反應熱見上述表3。
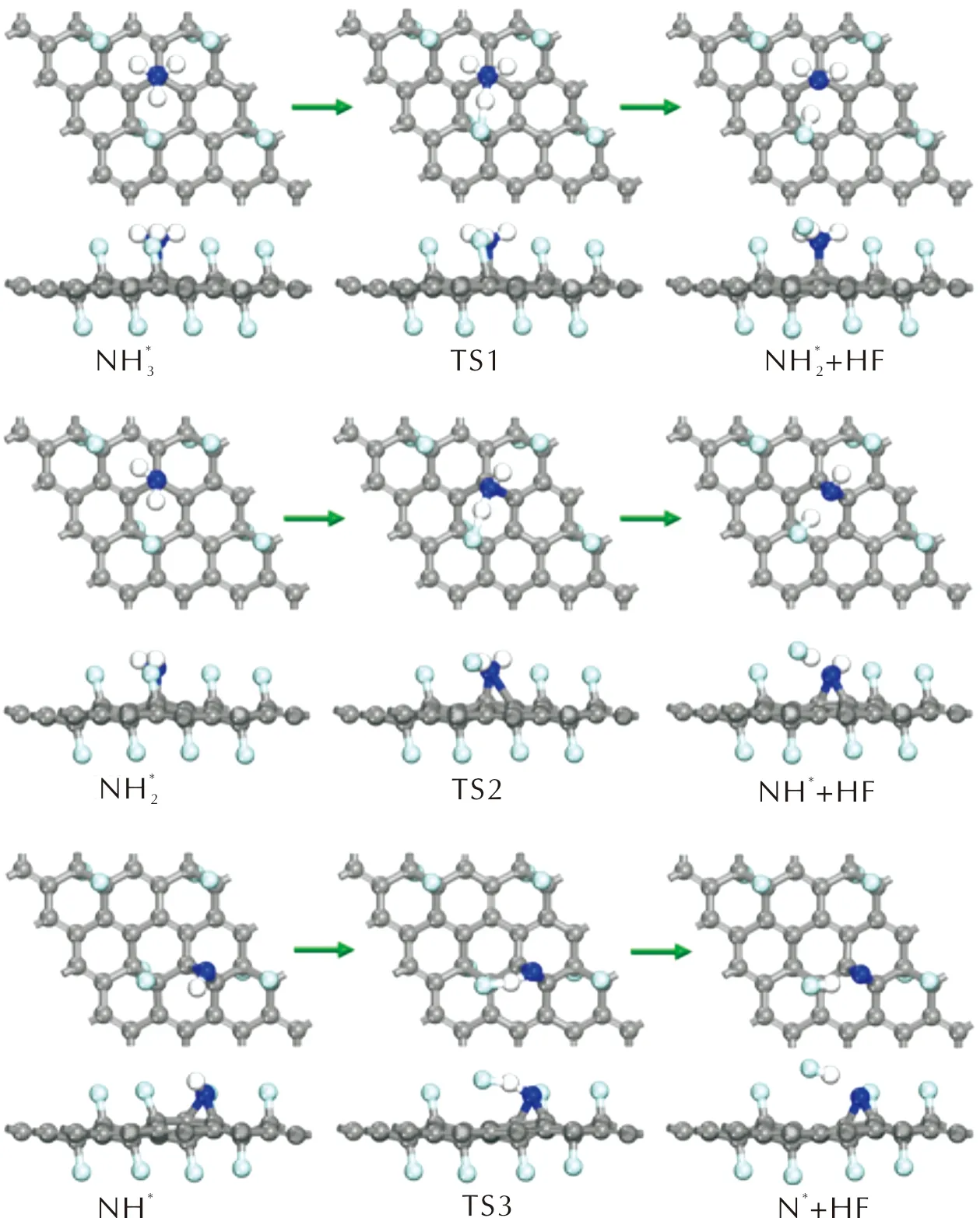
圖14 NH3在FG表面上生成HF分子的幾何結構Fig.14 The optimized structures of dehydrogenation reactions of NH3 to generate HF molecules on FG surfaces
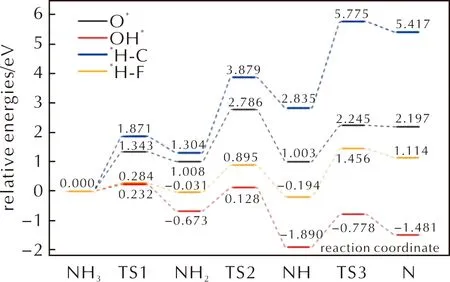
圖15 NH3分子在潔凈、氧化以及羥基化的FG表面上脫氫反應的能量曲線Fig.15 Energy curves of dehydrogenation reactions of NH3 on clean,oxidized,and hydroxylated FG surfaces
當NHx在潔凈的FG表面與F原子反應生成HF分子時,反應的活化能僅略高于NHx與OH*基團反應的活化能,并且遠低于O*存在時的活化能。因此NH3在FG表面將首先與F原子反應生成HF分子。同時由于NH3分子脫氫生成HF分子的反應活化能遠低于HClO4分子分解路徑上的控速步驟活化能。因此當AP在FG表面反應時,NH3分子會比HClO4分子更迅速地大量分解并生成HF分子,而不是在HClO4分子分解一定程度后才開始大量分解。這有助于AP分解過程中產生的游離NH3分子的消耗,從而防止游離的NH3吸附在AP表面。同時NH3分子的分解會降低FG表面的F原子覆蓋度,導致HClO4分子在F原子覆蓋度降低的FG表面進行反應。
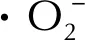
3 結 論
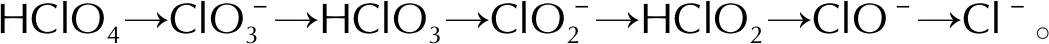
(2) HClO4分子分解過程中,會不斷分解出O*原子以及OH*基團吸附于FG表面。OH*基團會參與到HClO4分子的分解過程中,并通過反應生成O*原子。O*原子的吸附不利于HClO4分子在FG表面的分解。
(3) AP分解產生的另一初級產物NH3分子及其脫氫產物能夠與FG表面的O*原子反應生成OH*,同時其也易于與OH*反應生成H2O分子,從而降低FG表面的O*原子的覆蓋度。即NH3分子在FG表面的脫氫反應促進了FG表面O*覆蓋度的降低,因而有利于HClO4分子的分解。
(4) FG催化劑表面的F原子同樣易于與NH3反應,從而消耗AP分解過程中產生的游離的NH3分子,防止NH3分子吸附在AP表面。AP分解過程中阻滯期的形成主要是因為AP分解生成的NH3分子在AP表面的吸附,因此FG催化劑能夠有效地消除AP分解的阻滯期。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
