固相萃取/超高效液相色譜-串聯(lián)質(zhì)譜法測定兒童化妝品中新康唑等4種抗生素
2023-01-05 11:16:30聶明霞梁文耀李鑫宇文嘉林夏澤敏譚建華
分析測試學報 2022年12期
汪 毅,聶明霞,賈 芳,梁文耀,李鑫宇,文嘉林,夏澤敏,譚建華
(廣州質(zhì)量監(jiān)督檢測研究院,國家化妝品質(zhì)量檢驗檢測中心,廣東 廣州 511447)
近年來,關于兒童化妝品中激素、抗生素等藥物非法添加的報道頻出,兒童化妝品的質(zhì)量安全問題已成為大眾的關注焦點。相關研究表明,不規(guī)范使用抗生素類藥物容易造成過敏性皮炎、肝功能損傷等不良影響,并使人體產(chǎn)生耐藥性[1-3],其中兒童是抗生素濫用中的最大受害者[4-6]。筆者在使用高分辨質(zhì)譜篩查化妝品中禁用物質(zhì)的日常工作中發(fā)現(xiàn),市場上一些兒童化妝品(主要是嬰幼兒護膚膏)存在新康唑等抗生素非法添加情況。新康唑等并不在此前的一些化妝品相關規(guī)范和標準[7]規(guī)定的抗生素檢驗方法范圍內(nèi),雖然目前一些藥物或土壤中此類化合物的測定方法已有文獻報道[8-9],但化妝品與這些基質(zhì)差異較大,已有方法無法直接應用于化妝品中相關化合物的測定。因此亟需建立高靈敏、快速且能同時分析新康唑等多種組分的測定方法[10],為相關監(jiān)管工作提供技術支撐。
液相色譜-質(zhì)譜聯(lián)用技術是抗生素分析檢測的有效技術手段[11-13],但由于許多樣品在分析過程中存在基質(zhì)抑制現(xiàn)象,因此需要進一步凈化。新康唑、硫康唑、抑霉唑、噻康唑是咪唑類衍生物[14],脂溶性較強,且此類化合物咪唑環(huán)上的吡啶N堿性較強,因此本文使用強陽離子交換SPE小柱去除雜質(zhì),消除基質(zhì)抑制,使目標物獲得了良好回收率。本文建立的化妝品中新康唑等4種禁用物質(zhì)[7]的超高效液相色譜-串聯(lián)質(zhì)譜檢測方法,將有效填補抗生素檢驗方法的缺口,對保障消費者特別是兒童群體的權益和健康具有重大意義。
1 實驗部分
1.1 儀器與試劑
島津液相色譜儀-SCIEX AB 5500+三重四極桿質(zhì)譜儀(日本島津與美國SCIEX公司);BSA224SCW電子天平(德國賽多利斯公司);MS3 basic渦旋振蕩器(德國IKA公司);KQ-250DV型數(shù)控超聲波清洗儀(昆山市超聲儀器有限公司);Milli-Q純水系統(tǒng)(美國Millipore公司);固相萃取系統(tǒng)(上海安譜科學儀器有限公司);強陽離子交換小柱(200 mg,3 mL,上海安譜科學儀器有限公司)。
抑霉唑(純度99.2%,CAS:35554-44-0,美國Dr.Ehrenstorfer公司);新康唑(純度98.0%,CAS:67914-69-6)、噻康唑(純度99.1%,CAS:65899-73-2)(美國CATO公司);硫康唑(純度95.0%,CAS:61318-90-9,美國Aladdin公司);甲醇、乙腈(色譜純,德國Merck公司);甲酸(分析純,上海安譜科學儀器有限公司);實驗用水(18.2 MΩ·cm)由Milli-Q純水系統(tǒng)制備。
1.2 標準溶液的配制
分別精密稱取4種對照品10 mg,使用乙腈溶解,配制成質(zhì)量濃度1 mg/mL的標準儲備溶液,4℃保存。分別取適量標準儲備溶液用70%乙腈稀釋,配制成10 mg/L的標準中間液。取適量標準中間液,用70%乙腈稀釋成0.1、0.2、0.5、2、5、10、20、50、100 μg/L的系列標準混合溶液。
1.3 樣品前處理
稱取樣品0.2 g(精確至0.001 g)于10 mL具塞比色管中,加入2 mL 70%乙腈水溶液(甲酸調(diào)至pH 3.0),渦旋30 s使樣品分散,再定容至刻度,渦旋30 s后,超聲提取15 min。取樣液6 mL于10 mL塑料離心管中,6 500 r/min離心10 min。
取強陽離子交換固相萃取小柱,分別用5 mL甲醇和10 mL超純水活化,取離心上清液4 mL加入固相萃取柱,使其自然流下,用4 mL 2%甲酸水淋洗柱床后,再用4 mL含10%異丙醇的乙腈溶液淋洗柱床,洗耳球擠干后用4 mL含10%異丙醇的乙腈溶液(含2%氨水)洗脫目標物,收集流出液,過0.22 μm濾膜后待測。
1.4 儀器分析條件
色譜條件:Waters ACQUITY BEH C18柱(2.1 mm×100 mm,2.5 μm);流動相:A相:0.1%甲酸水,B相:乙腈;流速:0.3 mL/min;進樣量:3 μL;柱溫:40℃;梯度洗脫程序:0~7 min,20%~50%B;7~9 min,50%B;9~9.1 min,50%~90%B;9.1~12 min,90%B。
質(zhì)譜條件:離子源為電噴霧離子源(ESI源);監(jiān)測模式為正離子多反應監(jiān)測(MRM);離子化電壓4 500 V;氣簾氣壓力137.8 kPa;噴霧氣壓力344.7 kPa;碰撞氣壓力62.1 kPa;離子源溫度450℃;監(jiān)測離子對、碰撞電壓(CE)和去簇電壓(DP)等參數(shù)見表1。

表1 4種抗生素的質(zhì)譜參數(shù)Table 1 Mass spectrometric parameters of 4 antibiotics
2 結果與討論
2.1 方法建立
2.1.1 色譜柱的選擇4種抗生素的logP值在3.58~5.66之間,在C18色譜柱上均有較好的保留,因此考察了Waters ACQUITY HSS C18(2.1 mm×100 mm,1.8 μm)、Waters ACQUITY BEH C18(2.1 mm×100 mm,2.5 μm)、Agilent Poroshell 120 SB C18(2.1 mm×100 mm,2.7 μm)、Phenomenex Kintext C18(3 mm×100 mm,2.6 μm)幾種不同品牌規(guī)格的C18色譜柱對化合物的分析效果。結果顯示各化合物在不同的C18色譜柱上均能得到有效分離,且在Waters ACQUITY BEH C18柱上的峰形更加尖銳對稱,因此用該柱進行后續(xù)分析。
2.1.2 流動相的選擇比較了水-乙腈、水-甲醇兩種流動相體系對4種抗生素的色譜分離效果。結果顯示,水-乙腈體系的洗脫能力更強,峰形更加尖銳。比較了10 mmol/L乙酸銨和0.1%甲酸兩種不同流動相添加劑對色譜峰的影響,發(fā)現(xiàn)流動相中添加0.1%的甲酸時,能通過降低pH值使化合物離子化,有效提高化合物的響應強度。因此,確定0.1%甲酸水-乙腈作為流動相。
2.1.3 質(zhì)譜條件的優(yōu)化在MS Only模式下,將200 μg/L的混合標準溶液通過針泵注入質(zhì)譜。調(diào)整質(zhì)譜條件和掃描模式,獲取化合物的母離子和子離子信息。在負離子模式下,4種化合物均無信號,而在正離子模式下能獲得信號強度較高的母離子。這是因為咪唑環(huán)上的吡啶N堿性較強,極易結合H+帶正電,因此使用正離子模式進行分析。通過優(yōu)化碰撞能、去簇電壓和離子源溫度等參數(shù),獲得強度較高且穩(wěn)定的化合物響應。
設置400、450、500、550℃4個離子源溫度,在正離子模式下對混合標準溶液進行分析,溫度升至450℃后,新康唑的信號強度有所降低,其他3種化合物響應強度幾乎無變化。因此,離子源溫度設置為450℃。
2.2 前處理方法優(yōu)化
2.2.1 提取溶劑的選擇乙腈水溶液對化妝品具有較好的分散、提取效果,通常用于化妝品中各類激素等物質(zhì)的測定[2,8]。本文以乙腈水溶液為提取溶劑,并比較了不同乙腈含量(10%、20%、30%、40%、50%、70%、90%)對4種抗生素的提取效果。如圖1所示,隨著乙腈含量從10%升高到90%,新康唑、硫康唑和噻康唑的回收率逐漸升高后降低:乙腈含量大于30%后化合物的回收率超過70%,當乙腈含量達到90%后,部分目標物的回收率低于20%;抑霉唑在各含量提取溶劑中的回收率均低于70%,達不到分析要求。可能原因是目標化合物脂溶性較強,使用高含量有機溶劑提取時,存在大量的防腐劑、表面活性劑等共提物,影響了目標物的離子化效率,產(chǎn)生嚴重的基質(zhì)效應。為此,本研究進一步對基質(zhì)效應進行確認。選取一陰性膏霜樣品0.2 g,使用10 mL 70%乙腈水超聲提取15 min,離心后取上清液加入一定體積的混合標準溶液(各目標物最終質(zhì)量濃度為10 μg/L),將相同濃度的溶劑混合標準溶液和基質(zhì)標準溶液上機分析。以基質(zhì)標液中化合物響應值/溶劑標液中化合物響應值計算基質(zhì)效應。實驗結果顯示,新康唑、硫康唑、抑霉唑、噻康唑的基質(zhì)效應分別為0.52、0.51、0.95、0.64,即化合物受到不同程度的基質(zhì)抑制。因此需要進一步對70%乙腈水提取的樣品進行凈化處理以獲得符合分析要求的回收率。

圖1 4種抗生素在不同含量乙腈中的提取回收率Fig.1 Extraction recoveries of 4 antibiotics in different proportions of acetonitrile
2.2.2 固相萃取小柱的選擇HLB固相萃取小柱的基質(zhì)是含有親脂性二乙烯苯的大孔共聚物,在pH 1.0~14.0范圍內(nèi)非常穩(wěn)定,對非極性至中等極性的酸性、中性、堿性化合物均有較好的回收率,適用范圍廣。考慮到目標化合物的脂溶性和弱堿性,使用HLB小柱對樣品進行凈化,將凈化后的樣液上機分析。結果顯示,新康唑的回收率低于50%,可能是因為HLB小柱不能將目標物與雜質(zhì)分離,未能有效消除基質(zhì)效應。同時,由于HLB小柱對目標物的反相保留太強,導致部分目標物難以從柱上完全洗脫,影響回收率,因此HLB小柱不適合用于4種抗生素的凈化。
SCX小柱的官能團為苯磺酸,提取目標物時磺酸基團顯示陽離子交換性質(zhì),而苯環(huán)顯示非極性吸附性質(zhì)。新康唑、硫康唑、抑霉唑、噻康唑為弱堿性化合物,pKa值分別為6.88、6.55、6.53和6.66,當溶液的pH值小于4.0時,化合物能夠完全離子化,與苯磺酸根結合,在柱上保留,用中性溶劑淋洗小柱即能去除中性和酸性共提物,達到凈化作用。因此使用SCX小柱凈化樣品。將凈化后的樣液上機測試,結果如圖2所示。經(jīng)SCX小柱凈化后,目標化合物的回收率較未凈化樣品顯著提高,新康唑的回收率更是從54.1%上升至96.3%。SCX小柱能有效凈化樣品,使化合物回收率滿足方法需求,因此選擇SCX小柱做進一步方法優(yōu)化。
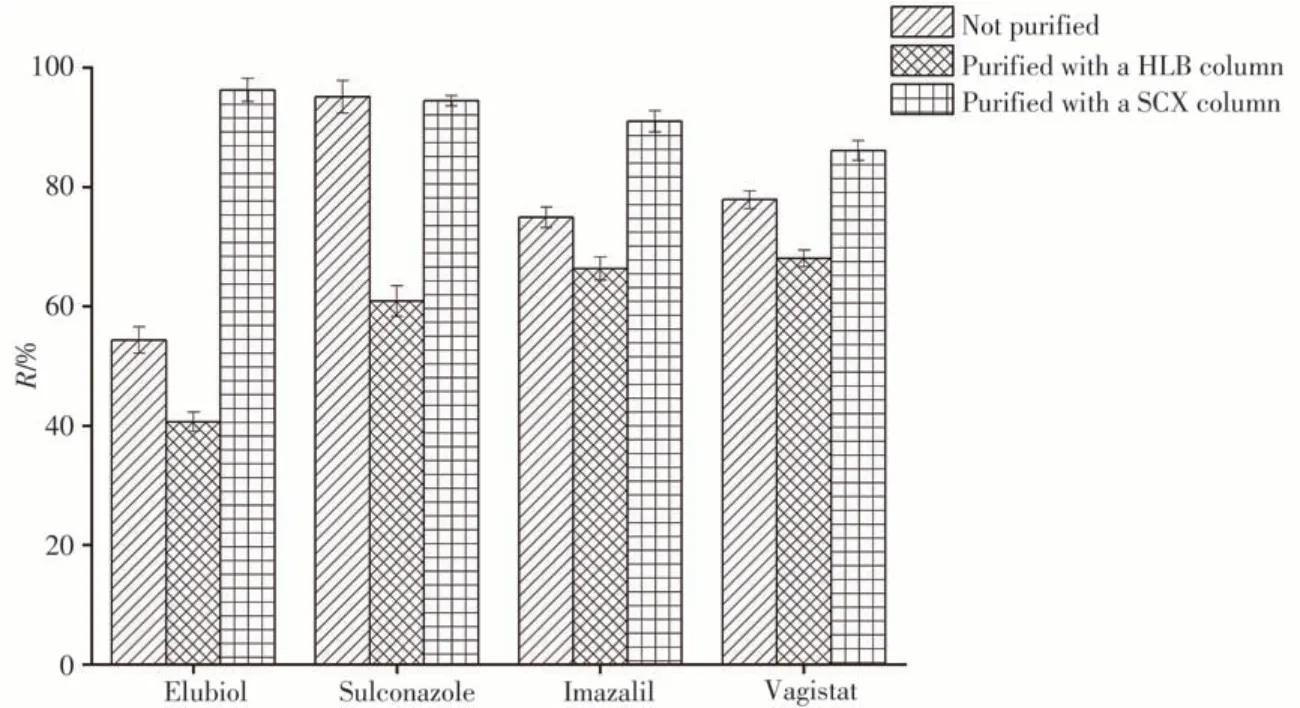
圖2 4種抗生素使用不同固相萃取小柱凈化后的提取回收率Fig.2 Extraction recoveries of 4 antibiotics purified with different solid phase extraction cartridges
2.2.3 洗脫溶劑的選擇采用4 mL含2%氨水的乙腈溶液對淋洗后的柱床進行洗脫,收集洗脫液上機測試。結果顯示新康唑、抑霉唑和噻康唑的回收率良好,硫康唑的回收率為75%,相對較低。繼續(xù)對柱床洗脫,第5 mL的洗脫溶液中仍有硫康唑檢出。可能是由于SXC小柱有陽離子交換和非極性吸附雙重作用,使得logP值較高的硫康唑難以洗脫,因此應選擇極性更低的洗脫溶液。使用4 mL含10%異丙醇的乙腈(2%氨水)溶液洗脫目標物,收集洗脫液上機測試,發(fā)現(xiàn)硫康唑的回收率提高至80%以上,因此選擇該溶劑為洗脫溶劑。
2.3 線性關系、檢出限及定量下限
配制系列混合標準溶液進行測定,以目標物的峰面積(Y)對相應的質(zhì)量濃度(X,μg/L)進行線性回歸,得到各化合物的標準曲線方程。結果表明4種抗生素在其質(zhì)量濃度范圍內(nèi)線性良好,相關系數(shù)(r)均為0.999。以3倍信噪比計算方法檢出限(LOD),以10倍信噪比計算方法定量下限(LOQ),得到4種抗生素的LOD和LOQ分別為0.005~0.250 μg/g和0.010~0.500 μg/g(表2)。結果表明,4種抗生素的線性關系良好,方法的檢出限和定量下限均可滿足化妝品的檢測需要。

表2 4種抗生素的線性關系、檢出限和定量下限Table 2 Linear relationships,LODs and LOQs of 4 antibiotics
2.4 回收率及相對標準偏差
選取3種典型化妝品(水、乳液、膏霜)作為加標基質(zhì),分別按照1倍、2倍和10倍定量下限進行低、中、高3個濃度水平的加標回收實驗,按照本方法進行前處理和測定,每個濃度水平平行測定6次,計算得到4種抗生素的平均回收率為89.3%~113%,相對標準偏差(RSD)為0.90%~7.2%(表3),滿足準確度和精密度要求。
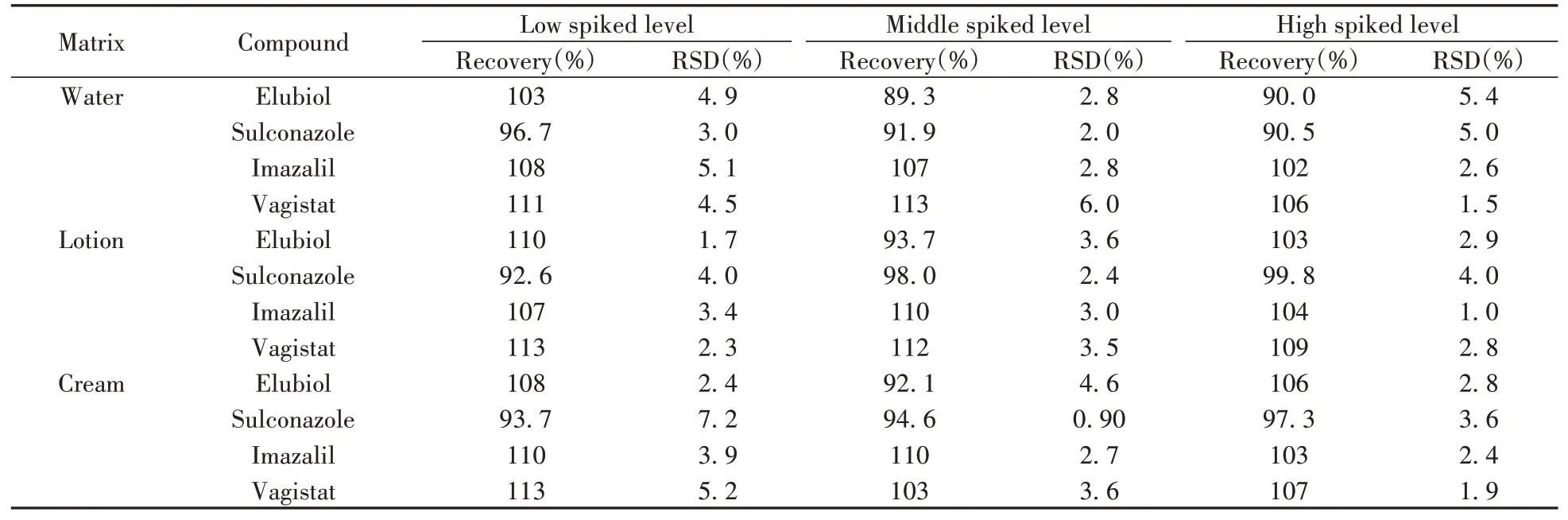
表3 4種抗生素的回收率和相對標準偏差(n=6)Table 3 Recoveries and RSDs of 4 antibiotics(n=6)
2.5 實際樣品測定
對市面上銷售的100多個批次的水、乳、膏霜使用上述方法進行測定,結果在4個批次產(chǎn)品中檢測出新康唑,含量范圍為0.24~1.25×104mg/kg,主要為兒童護臀霜產(chǎn)品。圖3為陽性樣品譜圖。

圖3 新康唑陽性樣品的提取離子色譜圖Fig.3 Extraction ion chromatogram of an elubiol positive sample
3 結論
通過優(yōu)化前處理條件和儀器方法建立了測定兒童化妝品中新康唑等4種抗生素的固相萃取/超高效液相色譜-串聯(lián)質(zhì)譜法,基于該類化合物咪唑環(huán)上吡啶N有較強堿性的特點,用強陽離子交換SPE小柱去除共提物,獲得了較好回收率。該方法高效、準確、特異性好,能夠為化妝品相關質(zhì)量監(jiān)管提供有效的技術支持。目前,雖然新康唑在兒童化妝品中存在非法添加事實,但缺少相關檢出情況的報告及檢驗方法,還處于監(jiān)管盲區(qū)。在此呼吁相關監(jiān)管部門擴大兒童化妝品中抗生素的監(jiān)測范圍,防止不法分子利用監(jiān)管漏洞非法牟利,危害兒童健康。
