Congenital nephrogenic diabetes insipidus arginine vasopressin receptor 2 gene mutation at new site: A case report
2023-01-04 07:59:44LuLuYangYanXuJianLiQiuQianYiZhaoManManLiHuiShi
World Journal of Clinical Cases 2022年36期
Lu-Lu Yang, Yan Xu, Jian-Li Qiu, Qian-Yi Zhao, Man-Man Li, Hui Shi
Lu-Lu Yang, Yan Xu, Man-Man Li, Hui Shi, Department of Pediatrics, Henan University of Chinese Medicine, Zhengzhou 450000, Henan Province, China
Jian-Li Qiu, Qian-Yi Zhao, Department of Pediatrics, The First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou 450000, Henan Province, China
Abstract BACKGROUND Congenital nephrogenic diabetes insipidus (CNDI) is a rare hereditary disorder. It is associated with mutations in the arginine vasopressin receptor 2 (AVPR2) gene and aquaporin 2 (AQP2) gene, and approximately 270 different mutation sites have been reported for AVPR2. Therefore, new mutations and new manifestations are crucial to complement the clinical deficiencies in the diagnosis of this disease. We report a case of a novel AVPR2 gene mutation locus and a new clinical manifestation.CASE SUMMARY We describe the case of a 48-d-old boy who presented with recurrent fever and diarrhea 5 d after birth. Laboratory tests showed electrolyte disturbances and low urine specific gravity, and imaging tests showed no abnormalities. Genetic testing revealed a novel X-linked recessive missense mutation, c.283 (exon 2) C>T (p.P95S). This mutation results in the substitution of a proline residue with a serine residue in the AVPR2 protein sequence. The diagnosis of CNDI was confirmed based on the AVPR2 gene mutation. The treatment strategy for this patient was divided into two stages, including physical cooling supplemented with appropriate amounts of water in the early stage and oral hydrochlorothiazide (1-2 mg/kg) after a clear diagnosis. After follow-up of one and a half years, the patient gradually improved.CONCLUSION AVPR2 gene mutations in new loci and new clinical symptoms help clinicians understand this disease and shorten the diagnosis cycle.
Key Words: Congenital nephrogenic diabetes insipidus; Arginine vasopressin receptor 2 gene mutation; New site; Diarrhea; Case report
INTRODUCTION
Congenital nephrogenic diabetes insipidus (CNDI) is a rare nephrogenic hereditary disorder. Approximately 90% of CNDI cases harbor arginine vasopressin receptor 2 (AVPR2) mutations transmitted by Xlinked recessive inheritance. Less than 10% of CNDI cases harbor aquaporin 2 (AQP2) mutations that are autosomal recessive or dominant mutations, and the genetic causes are unknown in approximately 2% of CNDI cases. To date, approximately 290 AVPR2 gene variants that may cause CNDI have been reported in the Human Gene Mutation Database[1], including approximately 177 missense mutations[2]. The disease often develops in infancy and early childhood, during which growth retardation, frequent vomiting, and hyperthermia represent the more common symptoms[1]. Growth retardation is considered a serious complication of CNDI. In addition to urinary complications, short stature and CKD are common[3]. Intracranial calcification and epilepsy are rare complications. New mutation sites and new clinical manifestations are constantly being reported. In this case, a pediatric patient with CNDI and a AVPR2 gene mutation at a new locus is reported. In addition, the diarrhea observed in this case is likely related to the AVPR2 gene mutation at the new locus. We discussed the new mutation and new phenotype together with information reported in the literature to improve clinicians’ understanding of CNDI and guide the clinical diagnosis of this disease.
CASE PRESENTATION
Chief complaints
A 48-d-old Chinese boy presented with fever and diarrhea for one month.
History of present illness
A 48-d-old boy presented with recurrent fever and diarrhea 5 d after birth. In the following month, the child developed repeated fever, diarrhea, and a body temperature fluctuating between 37.1 °C and 38.4 °C. The fever was irregular, and the diagnosis could not be confirmed after 3 hospitalizations at the local hospital. To further confirm the diagnosis, the child was admitted to our hospital for treatment, and the admission symptoms included clear consciousness, general spirit, fever, diarrhea, no dehydration, sunken nose bridge, dry skin, normal diet, normal sleep, and normal urination.
History of past illness
The patient had no previous medical history.
Personal and family history
The patient’s parents deny any family history.
Physical examination
The patient’s temperature was 38.1 °C, pulse 140 beats/min, respiratory rate 35 breaths/min, blood pressure 70/40 mmHg, and weight 5 kg. No abnormalities were found in other system examinations.
Laboratory examinations
Blood test analysis showed an electrolyte imbalance: K, 5.5 mmol/L; Na, 149.5 mmol/L; Cl, 112.6 mmol/L; Ca, 2.61 mmol/L; and P, 1.98 mmol/L. Urinalysis showed that the specific gravity of urine was less than 1.005. Routine blood cell count, erythrocyte sedimentation rate, routine stool examination, 10 items regarding prenatal and postnatal care, 6 items regarding immunity, tuberculosis antibody test, Epstein-Barr virus nucleic acid quantitative detection, cytomegalovirus nucleic acid quantitative detection, infectious disease screening, genetic testing, glucose monitoring test and other results were normal.
Imaging examinations
Color Doppler ultrasound of the superficial lymph node, lung, heart, gastrointestinal tract, liver, gallbladder, spleen, abdominal lymph node and kidney revealed normal findings.
Genetic examinations
An AVPR2 gene mutation, c.283 (exon2) C>T (p.P95S), was identified. This mutation was a hemizygous mutation in the 2ndexon of the AVPR2 gene. The C nucleotide at position 283 became a T nucleotide, which led to an amino acid change. The amino acid at position 95 was mutated from proline to serine [p.Pro95Ser (p.P95S)]. This mutation was a missense mutation and a novel mutation locus in the AVPR2 gene. The mother of the proband is a carrier of this gene mutation, and the father harbors no abnormal mutations in this gene. The specific genetic test results and family pedigree are shown in Figures 1 and 2. The test results showed that the proband had a hemizygous mutation, which was consistent with X chromosome recessive inheritance. Because the child had intractable diarrhea and a possible pathogenic variant of diarrhea could not be excluded, we tested related genes. No abnormalities in JAK3 (severe combined immunodeficiency, autosomal recessive, T-B+NK-), ZAP70 (selective T-cell deficiency) or PLEC (epidermolysis bullosa and pyloric atresia) were noted in the genetic testing.
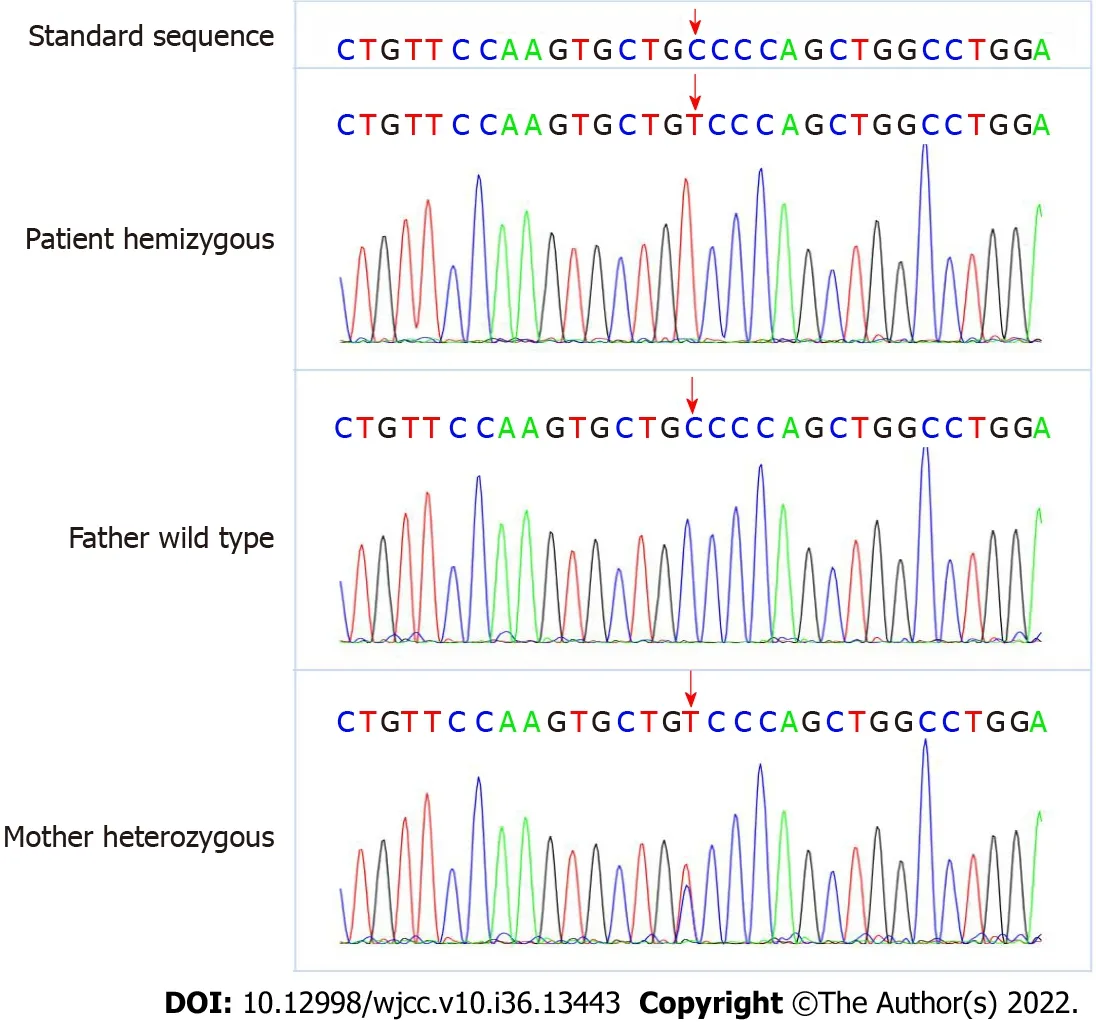
Figure 1 Genetic testing results of the child indicated a arginine vasopressin receptor 2 gene mutation, c.283 (exon2) C>T (p. P95S). This was a hemizygous mutation in the 2nd exon of the arginine vasopressin receptor 2 (AVPR2) gene. The base C at position 283 became base T, which led to the change of amino acid. The amino acid at position 95 was mutated from proline to serine, p. P95S (p. Pro95Ser). This mutation was a missense mutation and a new mutation site in the AVPR2 gene. The mother of the proband is a carrier of this gene mutation, and the father has no abnormal mutation in this gene.

Figure 2 Family pedigree of patients.
FINAL DIAGNOSIS
Based on the clinical symptoms of fever and diarrhea at admission, we performed auxiliary examinations to clarify the diagnosis and exclude related diseases. Furthermore, a genetic diagnosis of a c.283 (exon 2) C>T (p.P95S) mutation of the AVPR2 gene is of great significance in the diagnosis of CNDI, and the reference standards are stated here: (1) CNDI is a rare inherited disease that is caused by mutations in AVPR2 or AQP2[1]; (2) Mutations in either AVPR2 or AQP2 result in a genetic disease known as nephrogenic diabetes insipidus[4]; (3) CNDI results from mutations in the AVPR2 or AQP2 genes[5]; and (4) AVPR2 mutations result in X-linked recessive NDI, the most common form of inherited NDI[6]. As a result, the patient was finally diagnosed with CNDI.
TREATMENT
After the child was admitted to the hospital, we did not confirm the diagnosis in a timely manner. Treatment during this period included physical cooling supplemented with appropriate amounts of water. However, this approach does not fundamentally solve the problem. Two weeks after discharge from the hospital, we confirmed the diagnosis based on the results of genetic testing. Oral hydrochlorothiazide (1-2 mg/kg based on the child's body weight) was administered twice a day. After taking the medication for one year, the doctor adjusted the medication according to the child’s physical condition and took it orally thereafter (1-2 mg/kg, twice a day). Tables 1 and 2 present the changes in the condition of the child before and after the oral administration of hydrochlorothiazide.

Table 1 Clinical presentations of the child before oral administration of hydrochlorothiazide

Table 2 Clinical presentation of the child after oral administration of hydrochlorothiazide
OUTCOME AND FOLLOW-UP
The child attended follow-up at the outpatient clinic after discharge, and the family members were instructed to monitor the changes in the child’s body temperature, stool, diet and height. The follow-up period lasted approximately one and a half years. The child’s condition is relatively stable now with 1400-1600 mL/d water intake, and other aspects of growth and development are similar to that noted in normal children.
DISCUSSION
In 1992, van den Ouwelandet al[7] reported for the first time that patients with X-linked recessive nephropathy insipidus harbor AVPR2 gene mutations and gradually began to study the genetics of CNDI at home and abroad. CNDI patients show symptoms of polyuria and polydipsia from birth and typically develop irritability[8], feeding difficulties, weight loss, dry skin, poor skin elasticity, sunken eyes and other dehydration manifestations in the first week after birth. High fever and constipation are frequently noted[9,10]. Common symptoms in male patients include polyuria, polydipsia, fever of unknown etiology, convulsions, and vomiting. These symptoms typically appear shortly after birth, and relatively mild symptoms are generally noted in women[11]. In this case, the child also had symptoms of long-term recurrent fever. Schrageret al[12] reported on fever in CNDI patients but did not further clarify the mechanism of the fever. Some researchers believe that this intermittent high fever is a common complication of the dehydration state, which mainly occurs in very young children[9]. Based on the fact that the fever can be further relieved after the patient drinks water, some scholars consider it to be a dehydration fever[13]. In addition to the fever symptoms, the child we described also had obvious symptoms of diarrhea. In this regard, we hypothesized that the patient’s repeated fever is caused by dehydration as a result of diarrhea. It is well known that the main symptom of CNDI patients is polyuria[5,14-16]. To date, there have been no reports of diarrhea associated with this disease. Genetic testing of the AVPR2 gene revealed the c.283 (exon 2) C>T (p.P95S) mutation, which was considered to be associated with diarrhea symptoms in this patient. This presentation potentially represents another new complication of the disease. This new clinical manifestation potentially results from this novel mutation locus.
Research on the pathogenesis of CNDI has been previously reported in detail. Under physiological conditions, AVP secreted by the posterior pituitary increases in the presence of hypovolemia or hypernatremia, which binds to the type 2 receptor AVPR2 located on the basolateral side of collecting duct cells[9], and activated AVPR2 initiates a signal transduction cascade. This process includes the activation of adenylate cyclase by stimulating Gs protein[4], resulting in increased intracellular cyclic adenosine monophosphate (cAMP) levels, activation of protein kinase A (PKA), and phosphorylation of AQP2. The phosphorylation of three monomers in the AQP2 tetramer results in the redistribution of the AQP2 homotetramer, transportation of AQP2 from storage vesicles to the apical membrane, and increased permeability of collecting duct chief cells to water[9,17]. The important regulatory role of AVPR2 can be clarified from the pathogenesis process. This mechanism also represents the theoretical basis for direct sequencing of AVPR2 in neonates with familial susceptibility to CNDI as a rapid diagnostic tool for CNDI in clinical practice[14].
According to classification standards, researchers have categorized AVPR2 gene mutations into three categories: Type I mutants result in proteins that reach the cell surface but cannot bind to their ligands, type II mutant receptors are damaged and cannot reach the cell surface, and type III mutant proteins are improperly transcribed[11]. The diarrhea in this case likely resulted from the AVPR2 gene c.283 (exon 2) C>T hemizygous mutation, which caused the substitution of proline at position 95 to serine. This substituted based is surrounded by leucine at position 94 and leucine at position 96. Glutamine reacts to form new hydrogen bonds. The formation of new amino acid sequences leads to improper folding of the AVPR2 along with protein conformation changes. Thus, the protein is retained in the intracellular endoplasmic reticulum (ER)[5,15]. This effect may alter some properties of the protein, causing it to become harmful[1]. The accumulation of this harmful product affects the intestinal mucosa or intestinal muscle layer of infants and young children through a specific channel or by releasing a specific factor. These children have weak intestinal function and are easily affected; thus, clinical symptoms of diarrhea will occur. Alternatively, the formation of new amino acid sequences leads to misfolding of the AVPR2, which enables the rapid degradation of certain peptides[15] and may also produce harmful proteins that affect the body’s brain-gut axis function. Another hypothesis is that mutations in the AVPR2 gene do not cause alterations in expression, ER retention, or constitutive endocytosis. Rather, the symptoms are due to the abnormal function of the mutant receptor[1]. The function of the protein may cause intestinal flora disturbances. Currently, this is only a hypothesis for the mechanism by which diarrhea symptoms develop, and further studies involving a definitive functional analysis of this mutation are needed. In this study, the c.283 (exon2) C>T (p.P95S) mutation in the AVPR2 gene and the symptoms of diarrhea are reported. However, the effect on protein structure and function is unclear. The next step is to analyze the function of the receptor to verify the specific impact of the mutation on the receptor, provide a theoretical basis for emerging clinical complications, and propose possible treatment strategies for specific functional defect[1].
CNDI is a severe form of DI that is difficult to treat and typically results from genetic defects[15]. Currently, there is no specific treatment method for CNDI, and treatment mainly focuses on improving symptoms in clinical practice, such as guiding patients to supplement sufficient fluids, a low-sodium diet, a low-protein diet, and the oral administration of some drugs[5,18]. Based on advancements in molecular biology technology, some new therapeutic methods exhibit great potential in treating children with CNDI. Currently, these methods mainly involve the following mechanisms: (1) AVPR2 antagonists and cell-permeable AVPR2 antagonists act as molecular chaperones and are mainly administered to patients with AVPR2 missense mutations[19]. Targeting chemical and molecular chaperones appropriately corrects the point mutations that cause misfolded disease-causing proteins, rescuing mutant proteins from ER retention and allowing correctly folded proteins to be delivered in cells[19,20]; (2) AVPR2 agonists, some of which are cell permeability agonists, can bind AVPR2 mutants trapped in the ER, but they do not stabilize their conformation and can directly activate AVPR2 mutants within these cells by signaling to transmit a preformed receptor-G protein-adenylate cyclase complex. The subsequent generation of cAMP activates PKA, resulting in AQP2 phosphorylation and plasma membrane expression, thereby attenuating the NDI phenotype[4,19]; (3) Some classic treatment modalities, including vasopressin analogs, prostaglandin receptor agonists, hormone receptor agonists, and cGMP phosphodiesterase inhibitors, can bypass the defective AVPR2 signaling pathway. The main mechanism of this therapeutic approach is the increase in cytoplasmic cAMP and activation of cAMPindependent pathways[4,18,19]; and (4) Gene therapy can be used to edit the genome of somatic cells or embryos to correct the mutated genes[19]. However, only theoretical studies have been performed at present due to ethical concerns related to this treatment method. Some newer potential drugs have also been reported, such as the AKAP-PKA interfering agent FMP-API-1, which is similar to vasopressin. FMP-API-1/27, a derivative of FMP-API-1, is the first low-molecular-weight compound found to phosphorylate AQP2 more efficiently than existing drug candidates. AKAP-PKA-interfering agents have the potential to be developed into new therapeutic drugs and become potential therapeutic options[21]. The AMPK activator NDI-5033 can improve the urine concentration of NDI animals and is expected to be a potential therapy for CNDI caused by AVPR2 mutations[22]. Although numerous mutations with different functional defects hinder the development of specific treatments[23], the clinical safety, efficacy and long-term performance of these new potential treatments cannot be determined. With the development of technology and clinical safety evaluation improvements, these issues will be resolved in the near future.
CONCLUSION
This case describes a c.283 (exon 2) C>T (p.P95S) mutation of the AVPR2 gene in a child with CNDI with clinical symptoms of fever and diarrhea. Because children with CNDI do not exhibit the hallmark symptoms of chronic polyuria and polydipsia[14], the clinician experiences uncertainty in the early diagnosis process, which leads to a prolonged clinical diagnosis time. Many clinicians have considered CNDI when faced with infants with a fever of unknown etiology[10]. However, the onset of CNDI is insidious, and the condition may be misdiagnosed or missed. If the child has other rare or unreported phenotypes, the course of the disease lasts for a long time. Especially in neonates, CNDI can be lifethreatening because patients may develop severe dehydration, and repeated severe dehydration and hypernatremia can lead to intellectual disability[24]. This case broadens the genotype and phenotype spectrum of rare cases of CNDI caused by AVPR2 mutation and provides a basis for studying the molecular biology of AVPR2[25]. In addition, with research on the physiology and pathology of CNDI, the mechanism of these rare phenotypes will be understood.
FOOTNOTES
Author contributions:Yang LL reviewed the literature and contributed to manuscript drafting; Xu Y and Qiu JL reviewed the manuscript and guided its revision; Zhao QY participated in the clinical care of patients and collected data; Li MM and Shi H contributed to patient follow-up; Zhao QY and Qiu JL as the co-corresponding author of this manuscript; and all authors commented on previous versions of the manuscript and issued final approval for the version to be submitted.
Informed consent statement:Informed consent was obtained from the patient for the publication of this case report.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
CARE Checklist (2016) statement:The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Yan Xu 0000-0001-5258-339X; Jian-Li Qiu 0000-0001-6796-3774.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
 World Journal of Clinical Cases2022年36期
World Journal of Clinical Cases2022年36期
- World Journal of Clinical Cases的其它文章
- Precautions before starting tofacitinib in persons with rheumatoid arthritis
- Hoffa's fracture in a five-year-old child diagnosed and treated with the assistance of arthroscopy: A case report
- Development of dilated cardiomyopathy with a long latent period followed by viral fulminant myocarditis: A case report
- Short-term prone positioning for severe acute respiratory distress syndrome after cardiopulmonary bypass: A case report and literature review
- Compound heterozygous p.L483P and p.S310G mutations in GBA1 cause type 1 adult Gaucher disease: A case report
- Staphylococcus aureus bacteremia and infective endocarditis in a patient with epidermolytic hyperkeratosis: A case report