可見光/CuFeO2/H2 O2 體系降解氧氟沙星
2022-11-01 11:21:34張立東霍思月高孟春
蘭州交通大學學報 2022年5期
張立東,霍思月,李 杰,高孟春
(1.蘭州交通大學環境與市政工程學院,蘭州 730070;2.吉林化工學院資源與環境工程學院,吉林吉林 132022;3.中國海洋大學海洋環境與生態教育部重點實驗室,山東青島 266100)
氧氟沙星(OFX)屬于喹諾酮類抗生素,由于其具有較寬的抑菌譜、較強的活性和較少的不良反應被廣泛用于家畜疾病控制[1-2].近年來,OFX類抗生素的大量生產和過度濫用對生態環境產生了深遠的影響,并對人類健康構成一定威脅,因此,尋找先進的處理技術以降低OFX類抗生素的毒性,減輕其對生態環境和人類健康的威脅變得尤為重要[3].目前,抗生素類處理技術包括物理法[4]、生物法[5]和高級氧化技術(AOPs)[6].物理法主要包括膜分離和吸附,其成本較高且存在二次污染問題[4].生物法有好氧生物處理和厭氧生物處理等,但抗生素普遍存在生物毒性,易使微生物和細菌產生耐藥性[5].鑒于此,芬頓高級氧化技術受到人們廣泛關注,其能在電、光、催化劑等反應條件下產生高效活性氧物種(ROS),將大部分難降解有機物礦化成無機化合物或小分子物質進而達到高效去除污染物的目的[7].
傳統均相芬頓法具有降解效率高、工藝簡單和綠色環保等特點,主要原理是利用液相中的Fe2+和H2O2在酸性條件下反應產生高活性·OH氧化難降解有機物使污染物得以高效去除[8].然而此方法對pH值要求苛刻,還會產生大量的污泥[9].與之相比,異相類芬頓法使用固相鐵基催化劑代替溶解態Fe2+,具有pH適用范圍寬、二次污染少以及催化劑可重復利用的優勢[10].然而其反應過程中單一鐵礦物表面的Fe3+難以還原成Fe2+,導致催化活性低、穩定性差,因而實際應用受到限制[11].近年來,多金屬催化劑因具有多催化活性位點引起研究者的興趣.常見的多金屬固相催化劑有尖晶石型鐵氧體(MN2O4,其中:M、N為金屬)[12-14]、銅鐵礦型氧化物(MNO2)[15-16]和多金屬復合催化劑[17-19].研究人員發現將光催化技術與芬頓技術進行耦合時,光生電子可被Fe3+捕獲,將Fe3+還原為Fe2+,促使活性中心Fe2+得以再生,同時光生電子被Fe3+捕獲后促進了光生電子和空穴的分離,提高了光催化效率[20].然而,紫外光穩定性差、成本高,其在太陽能光譜中所占比例非常低,僅為4%.因此研發可見光響應的異相光芬頓催化劑至關重要[21-22].銅鐵礦 CuFeO2在地球中儲量豐富、環境友好且具有較窄的禁帶寬度,并展現出良好的光催化和芬頓活性[23],因而在難降解有機污染物處理方面顯示出良好的潛力.然而,利用傳統的固相燒結法和溶膠-凝膠法制備CuFeO2時需要較高溫度或大量使用有機溶劑,不僅制備成本高,還會在反應過程中產生大量雜質.
本文通過低溫水熱法制備高純度R-3m型CuFeO2催化劑,并通過各種表征手段對其結構及形貌等進行分析.以OFX作為目標污染物,考察所制CuFeO2催化劑在可見光下活化H2O2降解OFX的效能、機制和循環穩定性.深入探究催化劑投加量、H2O2濃度、pH值對OFX降解效果的影響.利用紫外可見漫反射光譜、X射線光電子能譜分析CuFeO2的禁帶寬度和光學性能;通過自由基、光生電子和光生空穴捕獲實驗和電子自旋共振光譜鑒定可見光/CuFeO2/H2O2體系降解OFX的主要活性物質,并進一步揭示其降解機理.
1 材料與方法
1.1 異相催化劑CuFeO2制備
稱取2.4 g的 Cu(NO3)2·3H2O和 4.1 g Fe(NO3)3·9H2O于30 mL去離子水中,室溫下磁力攪拌30 min獲得完全溶解的黃綠色鹽溶液,加入0.5 g無水葡萄糖,待溶解后用0.5 mol·L-1的NaOH調節溶液的pH至11,最后將溶液緩慢倒入100 mL的不銹鋼反應釜中,于180℃下水熱反應24 h,待黑色產物冷卻至室溫后分別用去離子水和乙醇對其進行多次清洗,并于60~70℃下干燥,得到CuFeO2催化劑,并將其裝袋備用.
1.2 材料的測試表征
采用X-射線多晶衍射儀(XRD,D8 ADVANCE,German)對所制備CuFeO2的晶型結構進行分析,其中測試靶元素為Cu靶,Kα射線的波長為1.54?,測試范圍為10°~90°.采用掃描電子顯微鏡(SEM,JEOL-7500F,Japan)、能量色散 X射線能譜儀(EDS)以及透射電子顯微鏡(TEM,Tecnai G2 f20 s-twin)對CuFeO2的表面形貌以及表面尺寸結構特征進行分析.以Mg Kα射線(1 253.6 eV)作為激發源,采用Thermo Fisher Scientific公司所產X射線光電子能譜儀(XPS,ESCALAB 250,American)測定所制備CuFeO2的價帶譜.CuFeO2的光吸收性能以及禁帶寬度利用美國PE公司的紫外可見光譜儀(UV-Vis/DRS,Lambda 900,American)進行測定.
1.3 異相催化劑CuFeO2可見光下活化H 2 O2降解OFX的實驗
OFX的降解實驗采用外置光源型反應系統,主要由圓柱形的石英反應器、磁力攪拌器和300 W 的氙燈組成.反應器加入100 mL 10 mg·L-1的OFX溶液和一定劑量的CuFeO2催化劑.光芬頓催化降解反應前,先將反應器置于黑暗處攪拌1 h,達到吸附-解吸平衡,隨后開啟氙燈和攪拌器啟動催化反應.每間隔30 min取樣,采用日本島津儀器公司的L-2000高效液相色譜儀(HPLC)測定OFX的殘余濃度,HPLC采用Ultimate Plus-C18型色譜柱,尺寸為4.6 mm×150 mm×0.005 mm,柱溫為30℃,流動相為乙腈-水(V:V=60:40),流速為 1.0 mL·min-1,檢測波長為293 nm.采用電子自旋共振波譜儀(ESR,FA200)檢測降解過程中的活性自由基.分別利用式(1)和式(2)計算OFX的降解效率和擬一級動力學反應速率常數.
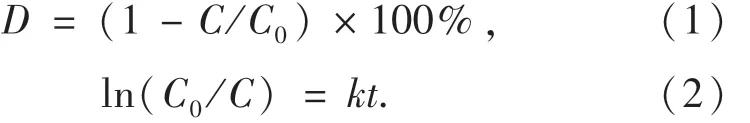
式中:D為OFX的降解效率;C0為OFX的初始濃度,mg·L-1;C為異相光芬頓降解時間為t時OFX的濃度,mg·L-1;k為一級動力學反應速率常數,min-1.
2 結果與討論
2.1 CuFeO2催化劑的表征
2.1.1 晶型結構和表面形貌
通過XRD、SEM、TEM和HRTEM等手段分析了所制CuFeO2催化劑的晶型結構和表面形貌,如圖1所示.由圖1(a)顯示,經過與 PDF#75-2416標準譜圖進行比對,所制催化劑在10°~80°出現的特征峰與R-3m相CuFeO2的峰型一一對應,反映3R型銅鐵礦CuFeO2制備成功.由圖1(b)的SEM圖可知,所制CuFeO2為斜方六面體結構,其表面光滑均勻、棱角分明,粒徑在 1~4μm 之間,不同粒度的CuFeO2顆粒團聚在一起,以團簇物的形式存在.圖1(c)所示的TEM圖也清晰展現了微米級CuFeO2的斜方六面體結構形態和團聚狀態.
2.1.2 能帶結構
為了分析p型半導體CuFeO2的能帶結構,對其紫外可見漫反射光譜和XPS價帶譜進行了測定.根據Kubelka-Munk公式(見式(3))繪制曲線如圖2所示.由計算可知,所制CuFeO2的禁帶寬度Eg值為2.73 eV.由 XPS價帶譜(見圖3(a))可知,CuFeO2的價帶位置為0.89 eV,故可得其導帶位置為-1.84 eV,由此畫出如圖3(b)所示的能帶結構圖.

圖 1 CuFeO2的 XRD圖、SEM 圖和 TEM 圖Fig.1 XRD,SEM and TEM patterns of CuFeO2
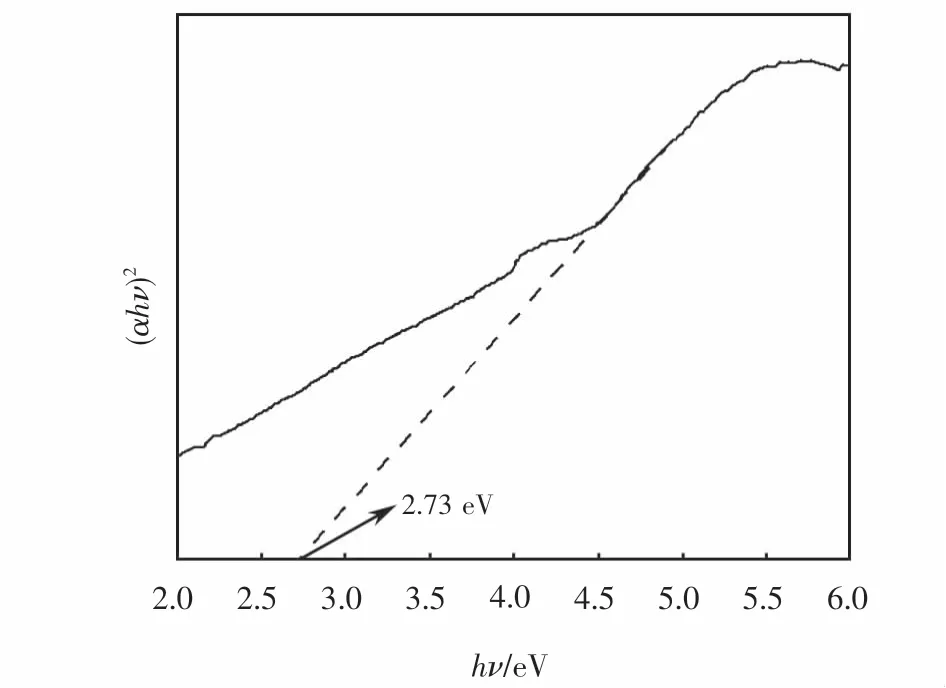
圖2 CuFeO2的紫外可見漫反射圖Fig.2 UV-Vis DRS of CuFeO2
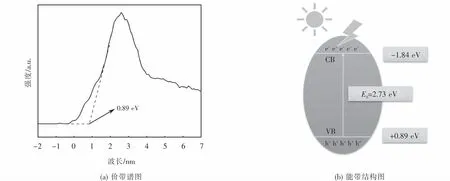
圖3 CuFeO2的價帶譜和可能的能帶結構Fig.3 Valence band XPS spectra and the possible band structures of CuFeO2
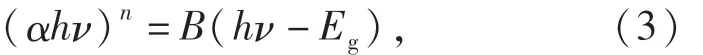
其中:h為普朗克常量;ν為光的頻率;B為常數;Eg為帶隙能量值;α為吸光度;n為1/2或者2(間接帶隙型半導體為1/2,直接帶隙型半導體為2).
2.2 可見光/CuFeO2/H 2O2 體系下降解 OFX的研究
2.2.1 不同體系下OFX的降解
為了考察可見光響應下所制CuFeO2活化H2O2對OFX的降解效果,分別在不同體系下進行了降解實驗并對相應的動力學行為進行了分析,降解條件如下:[H2O2]0=6 mmol·L-1,[CuFeO2]0=0.6 g·L-1,[OFX]0=10 mg·L-1.如圖4(a)所示,在未調節降解溶液初始pH的條件下,反應120 min后,單獨用6 mmol·L-1H2O2氧化降解10 mg·L-1OFX時,其去除效率僅為0.58%,這是由于H2O2的氧化還原電位值較低僅為1.77 eV,對有機污染物的氧化能力較弱[24];單獨可見光照射污染物時,其降解效率為20.2%,表明單獨可見光照射對OFX的去除效果不佳;CuFeO2催化劑對OFX的吸附曲線顯示,CuFeO2在降解1 h后達到吸附-解吸平衡,繼續延長吸附時間,由于CuFeO2的團聚,減少了CuFeO2顆粒與OFX分子在水中接觸的機會,因而OFX的濃度幾乎不再改變,CuFeO2對OFX的吸附率僅為19.9%;與單一體系相比,可見光/H2O2和可見光/耦合體系對OFX的降解效率分別增加25.4%和30.1%,這說明可見光對H2O2以及 CuFeO2均有一定的活化能力;當H2O2和CuFeO2耦合作用于污染物時,其降解效率為41.9%,這是因為CuFeO2能夠有效地活化H2O2產生自由基,促進OFX地降解;而本文所選可見光/CuFeO2/H2O2體系對OFX的降解效率高達85.3%,其較高的催化降解性能源于可見光和CuFeO2對H2O2的協同催化作用.此外,通過擬一級動力學方程進一步分析了OFX的降解過程.由圖4(b)可知,可見光/CuFeO2/H2O2體系降解 OFX的速率常數(1.48×10-2min-1)分別是 H2O2氧化(3.10×10-5min-1)和CuFeO2吸附(1.91×10-3min-1)的774.9倍和7.8倍,進一步說明CuFeO2可以作為一種有效的異相光芬頓催化劑活化H2O2降解OFX.
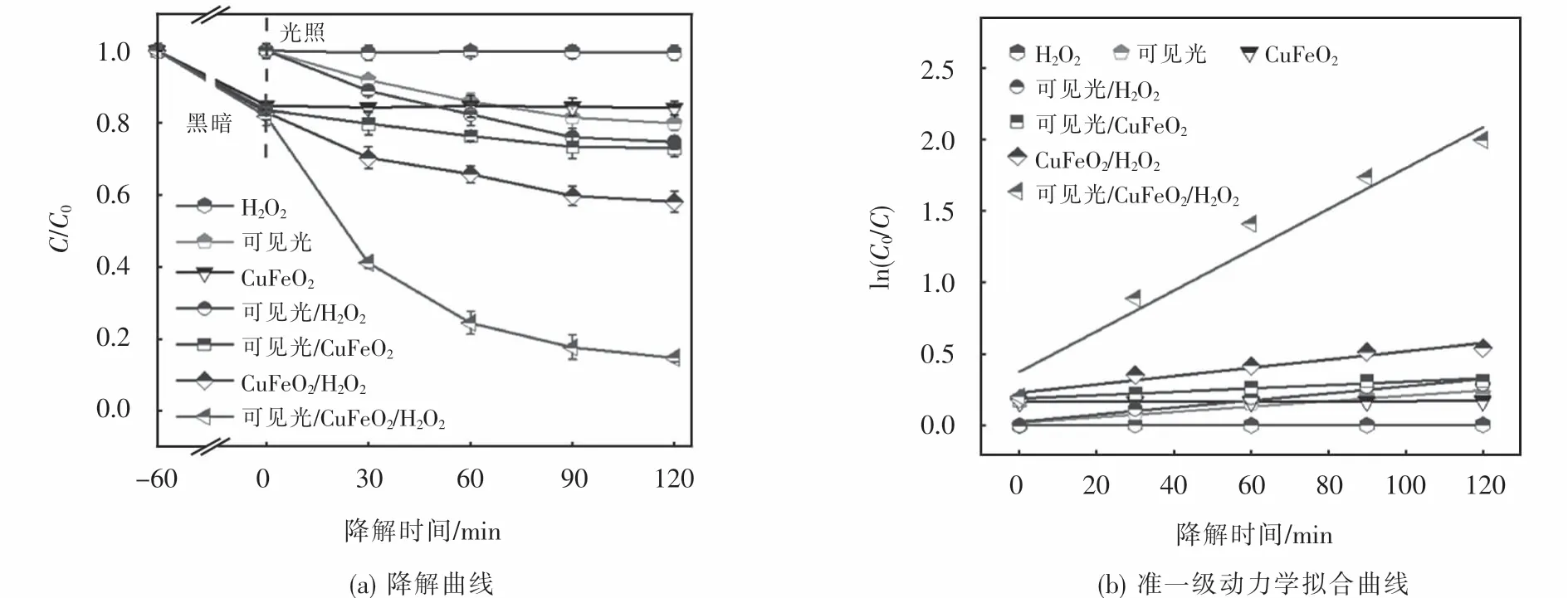
圖4 不同體系下OFX的降解曲線和準一級動力學擬合曲線Fig.4 Degradation and corresponding kinetic curves of OFX in various system
2.2.2 催化劑投加量和H2O2濃度對OFX降解的影響
在濃度為 10 mg·L-1的 OFX、10 mmol·L-1H2O2的條件下,通過改變CuFeO2催化劑的投加量,探究其在光芬頓體系下對OFX降解性能的影響,如圖5(a)所示,當CuFeO2催化劑的投加量從0.1 g·L-1增至 0.6 g·L-1時,可見光/CuFeO2/H2O2體系對OFX的降解效率由39.3%增至82.6%,這是由于隨著催化劑投加量地增加,吸附和催化H2O2的活性位點和光生載流子數目也隨之增加,促進了體系對OFX地降解.但是當CuFeO2催化劑投加量進一步增加至0.8 g·L-1時,OFX的去除效率由82.6%降為80.1%.這種現象是因為CuFeO2催化劑投加量地增加雖然會使活性位點和光生載流子數目增加,但受限于H2O2濃度,過量的催化劑活性位點無法得以良好的利用[25].此外,CuFeO2的過量投加還可能會造成溶液中活性自由基的不良消耗以及減弱可見光在體系中的透過性,對OFX的降解產生抑制作用.因此,0.6 g·L-1CuFeO2為異相光芬頓降解OFX的最佳催化劑投加量.
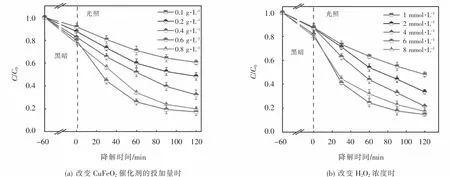
圖5 CuFeO2投加量、H 2 O2濃度對OFX降解的影響Fig.5 Effect of CuFeO2 dosage and H 2 O2 concentration on OFX degradation
在最佳CuFeO2催化劑投加量時,繼續探究H2O2濃度對可見光/CuFeO2/H2O2體系降解OFX的影響.由圖5(b)可知,當 H2O2濃度由1 mmol·L-1增至6 mmol·L-1,OFX的降解效率從56.4%增至85.2%,這是由于隨著H2O2濃度增加,體系中會產生更多的活性自由基,加速 OFX地降解.然而,將H2O2濃度進一步提高至8 mmol·L-1時,體系對OFX的去除效率下降至83.4%.上述結果表明,在一定范圍內提高H2O2濃度可以增加活性自由基的數量,然而過量的H2O2會通過式(4)和(5)發生副反應導致·OH的不良消耗,產生比·OH氧化能力更低的·O2H和·O-2[24],從而抑制了 OFX地降解.因此,異相光芬頓催化降解OFX的最佳H2O2濃度為6 mmol·L-1.
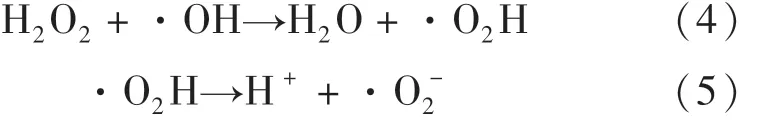
2.2.3 溶液初始pH對OFX降解性能的影響
在CuFeO2投加量為0.6 g·L-1和H2O2濃度為6 mmol·L-1條件下,考察了初始pH對可見光/CuFeO2/H2O2體系降解OFX的影響,其降解條件如下:[OFX]0=10 mg·L-1;[H2O2]0=6 mmol·L-1;[CuFeO2]0=0.6 g·L-1.由圖6可知,在未調節溶液初始pH(pH=3.6)時,經過120 min地降解,所制CuFeO2催化劑在可見光下活化H2O2對OFX的降解效率高達85.2%.當溶液pH調節至3,在降解時間為0~60 min時,OFX的降解效率明顯高于pH=3.6,這是因為在溶液pH為3時,體系中能夠產生足量的H+,使得可見光/CuFeO2/H2O2體系在短時間內產生大量活性自由基,從而促進OFX地降解.隨著降解反應地進行,溶液中積累的大量中間產物與OFX地降解產生競爭,導致OFX降解速率下降,其在120 min時的降解效率為85.7%,這與pH=3.6時的降解效率(85.2%)基本一致.然而當調節溶液初始pH為5、7和9時,OFX的降解率分別為76.7%、66.6%和47.4%,這是由于體系中pH的增加會產生高濃度OH-,從而加速了H2O2和活性自由基的不良消耗,使OFX的降解效率下降[26].綜上可知,在酸性體系下,CuFeO2催化劑在可見光下活化H2O2對OFX的降解效率最高,隨著pH的增加,可見光/CuFeO2/H2O2體系對OFX地降解產生抑制作用,盡管溶液pH為3時比未調節pH時OFX的降解速率更快,但在調節溶液pH過程中需要添加大量的酸,不僅過程繁瑣還會增加處理成本,因此,本文其他實驗均未調節溶液初始pH.
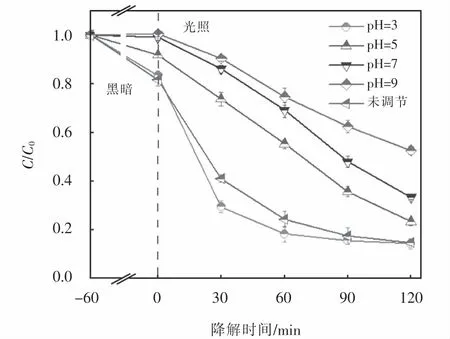
圖6 初始pH對OFX降解的影響Fig.6 Effect of initial pH on OFX degradation
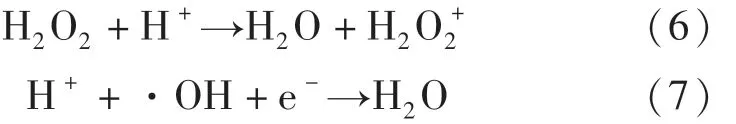
2.3 催化劑循環穩定性分析
為了評價可見光下CuFeO2活化H2O2的穩定性,在CuFeO2添加量為0.6 g·L-1,H2O2濃度為6 mmol·L-1和未調節溶液pH的條件下進行了連續5次催化降解OFX實驗.由圖7(a)可知,經過5次(連續600 min)的循環降解實驗,可見光/CuFeO2/H2O2體系對OFX的降解效率從最初的85.2%降低至77.5%,說明本研究所制CuFeO2作為異相催化劑在可見光下活化H2O2過程中具有良好的穩定性.通過分析5次催化降解反應前后CuFeO2的XRD譜圖的晶型結構,進一步探討了CuFeO2催化劑的穩定性.由圖7(b)可知,CuFeO2的XRD譜圖經過5次連續循環降解實驗幾乎沒有發生變化,其仍能保持原有晶型結構.上述結果證實,可見光/CuFeO2/H2O2體系降解OFX可以在較長時間內保持較高的催化活性.
圖7 可見光/CuFeO2/H 2 O2體系對OFX的5次循環降解,降解前后的XRD圖Fig.7 Five successive degradation cycles of CuFeO2/H 2 O2 system for the OFX degradation and XRD patterns before and after the degradation
2.4 CuFeO2催化劑Fenton體系下降解OFX機理
2.4.1 鑒定活性自由基
為了鑒定 CuFeO2可見光下活化 H2O2降解OFX的主要活性自由基,本研究分別采用甲醇、硝酸銀、對苯醌和草酸銨作為·OH、光生電子、·O2-和光生空穴的捕獲劑,其降解條件如下:[H2O2]0=6 mmol·L-1,[CuFeO2]0=0.6 g·L-1,[OFX]0=10 mg·L-1和[捕獲劑]0=50 mmol·L-1.由圖 8(a)可知,與不加捕獲劑的對照組相比,甲醇、疊氮化鈉、硝酸銀、對苯醌地添加明顯抑制了OFX地降解,而草酸銨地添加則對OFX降解沒有明顯影響.當添加甲醇、硝酸銀、對苯醌后,經過120 min的降解實驗,OFX的降解效率從對比組的85.2%分別下降到1.9%、53.2%、58.1%,而草酸銨添加后OFX的降解效率僅下降了3.1%.以上結果表明,·OH、光生電子、·O2-均在可見光/CuFeO2/H2O2體系降解OFX中起作用,而·OH是異相Fenton催化降解OFX的主要活性自由基.為了進一步研究可見光/CuFeO2/H2O2體系降解OFX生成活性自由基的情況,以二甲基吡啶N-氧化物(DMPO)為捕獲劑,分別在光催化體系(可見光/CuFeO2)、異相芬頓體系(CuFeO2/H2O2)和異相光芬頓體系(可見光/CuFeO2/H2O2)下進行ESR實驗.ESR譜圖(見圖8(b))顯示光催化體系內出現了極微弱的DMPO-·OH和DMPO-O-2信號峰,表明光催化體系無法產生·OH和·O-2,這與降解實驗結果相符合.而異相芬頓體系和異相光芬頓體系中均檢測到強度比為1:2:2:1的DMPO-·OH加合物的特征峰和強度比為1:1:1:1的DMPO-·O-2加合物的特征峰,表明CuFeO2/H2O2和可見光/CuFeO2/H2O2體系均可以產生·OH和·O-2.然而,在相同反應時間內,以上兩個體系DMPO-·OH特征峰的強度比DMPO-·O-2特征峰強度大,表明·OH是異相芬頓和異相光芬頓體系中的主要活性自由基,這與自由基捕獲實驗的結果一致.此外可見光/CuFeO2/H2O2體系特征峰的強度明顯高于CuFeO2/H2O2體系,這說明該體系下能夠產生的自由基濃度更高,這與圖4降解實驗相一致.
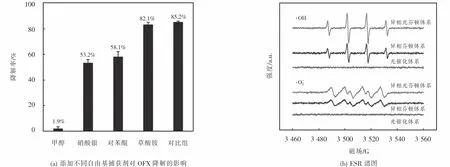
圖8 不同自由基捕獲劑對OFX降解的影響以及DMPO-·OH和DMPO-·O-2 的ESR譜圖Fig.8 Effect of various radical scavengers on OFX degradation and ESR spectra of DMPO-·OH and DMPO-·O-2
2.4.2 CuFeO2催化H2O2降解OFX的機理
基于自由基、光生電子和光生空穴捕獲實驗以及ESR分析結果,提出了如圖9所示CuFeO2可見光下活化H2O2降解OFX的可能機理,整個降解反應主要發生在CuFeO2表面的Cu和Fe活性位點上.當CuFeO2投加至污染物溶液中,一方面催化劑表面的Cu和Fe作為Lewis位點經過式(8)反應生成≡Cu+/Cu2+/Fe3+-OH絡合物,其中≡Cu+-OH可以通過式(9)與H2O2反應生成·OH和≡Cu2+-OH,生成的≡Cu2+-OH可經式(10)被H2O2還原成≡Cu+-OH并生成·O-2.與此同時,CuFeO2表面的≡Fe3+-OH也能與H2O2反應生成≡Fe2+-OH和·O-2(式(11)),而CuFeO2催化劑表面形成的≡Fe2+-OH又能通過式(12)催化 H2O2生成·OH 和≡Fe3+-OH.由于Cu2+/Cu+的氧化還原電位為 0.17 V,遠遠低于Fe3+/Fe2+的氧化還原電位值(0.77 V),因此 CuFeO2表面的≡Cu+-OH能將≡Fe3+-OH還原成≡Fe2+-OH(式(13)).通過催化劑表面的≡Cu+-OH/≡Cu2+-OH和≡Fe2+-OH/≡Fe3+-OH的氧化還原循環可以促進活化H2O2生成活性自由基.另一方面,經過可見光激發,CuFeO2可以通過式(14)產生光生電子和光生空穴.由圖3可知,CuFeO2的價帶位置為+0.89 eV,這比·OH/OH-的電勢(+2.4 eV)或·OH/H2O的電勢(+2.72 eV)更負,因此光生空穴不能將H2O氧化成·OH,而被激發產生的光生電子可以直接通過并將H2O2轉化成·OH(見式(15)).此外光生電子可以通過式(16)和式(17)促進催化劑表面Cu+和Fe2+的原位再生,極大地促進了活性自由基地產生.最終產生的·OH/·O-2可將OFX氧化成中間產物 CO2和 H2O(見式(18)).
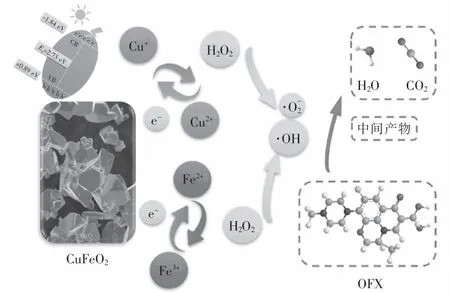
圖9 可見光/CuFeO2/H 2 O2體系中催化反應的機理示意圖Fig.9 Possible mechanism of OFX degradation by CuFeO2 in visible/CuFeO2/H 2 O2 system
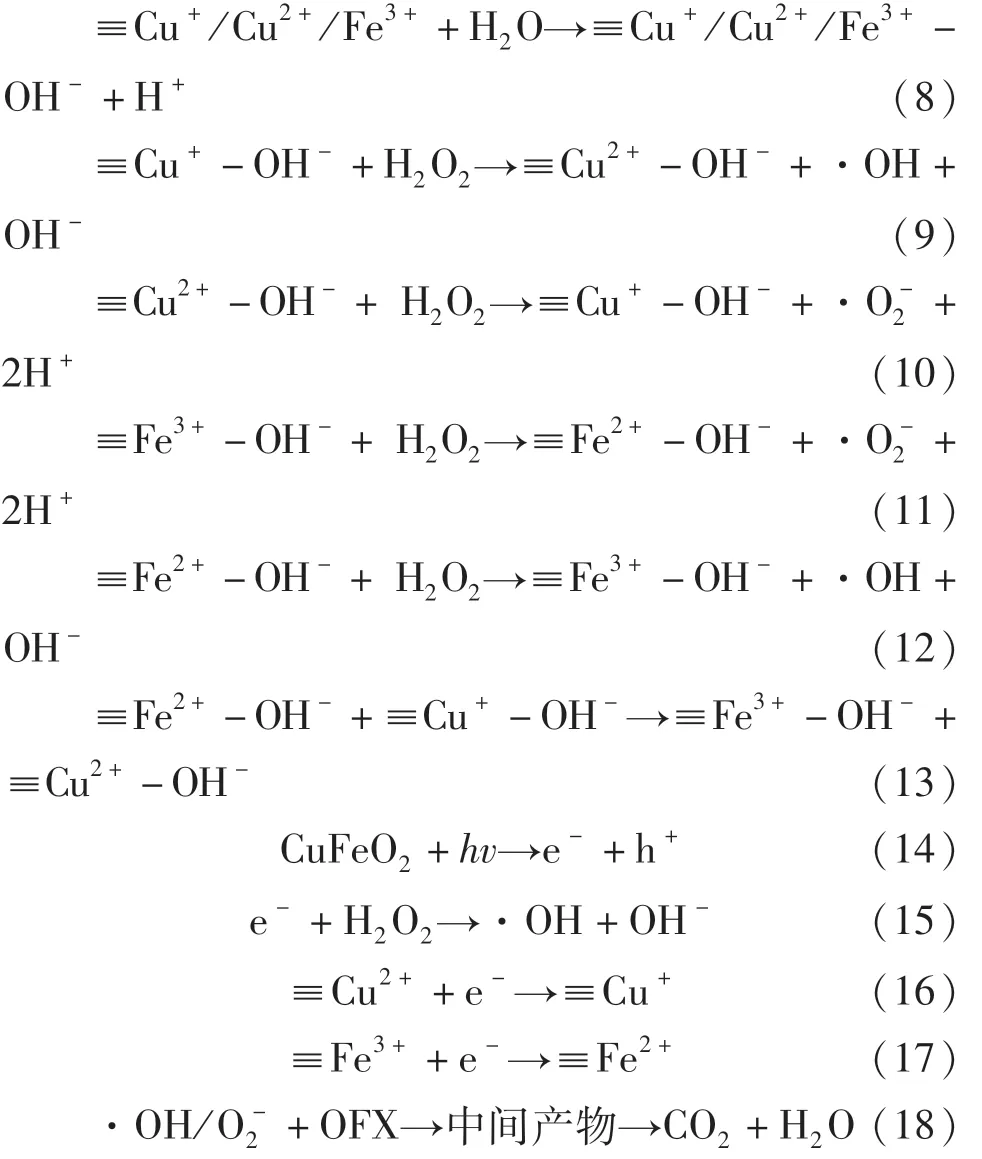
3 結論
1)通過水熱法成功制備高純度R-3m相結構CuFeO2催化劑,其表面光滑均勻、棱角分明,粒徑在1~4μm之間,且CuFeO2顆粒呈團聚狀態.
2)探究了可見光/CuFeO2/H2O2體系降解OFX的最佳降解參數,即催化劑濃度為0.6 g·L-1,H2O2摩爾濃度為6 mmol·L-1以及pH=3.6.在最佳降解條件下,CuFeO2催化劑在可見光下活化H2O2降解OFX的效率為85.2%.
3)經5次連續降解后,可見光/CuFeO2/H2O2體系降解OFX的效率僅降低了7.7%;XRD譜圖證實降解反應后CuFeO2仍能保持原有的晶型結構,結果表明所制CuFeO2具有良好的穩定性.
4)自由基、光生電子和光生空穴捕獲實驗以及ESR測試結果表明可見光/CuFeO2/H2O2體系中·OH為主要活性自由基;提出了可能的降解機理,揭示了CuFeO2中的Fe和Cu雙活性位點能夠加速Fe3+/Fe2+和Cu+/Cu2+的氧化還原循環,促進活性自由基地生成,加速OFX地降解.
猜你喜歡
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
甘肅教育(2020年14期)2020-09-11 07:57:42
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國衛生(2014年11期)2014-11-12 13:11:32
新高考·高一物理(2014年1期)2014-09-18 01:26:07
體育師友(2011年2期)2011-03-20 15:29:29
