超微粉碎和高壓均質聯合處理對幾丁質理化性質及微觀結構的影響
2022-10-28 07:17:52馬愛進桑亞新孫紀錄
食品科學 2022年19期
劉 洋,肖 宇,馬愛進,桑亞新,孫紀錄,*
(1.河北農業大學食品科技學院,河北 保定 071001;2.北京工商大學食品與健康學院,北京 100048)
幾丁質是自然界中僅次于纖維素的第二大豐富的多糖,由-1,4-糖苷鍵連接的-乙酰基氨基葡萄糖組成,廣泛分布于真菌的細胞壁、無脊椎動物和甲殼類動物的外骨骼中,其中甲殼類動物是幾丁質的主要來源。近年來,蝦、蟹等甲殼類動物的加工和消費量迅速增長,產生了大量的蝦蟹殼等副產物,為幾丁質的生產提供了豐富的原料。幾丁質生物活性的不斷發掘推動了幾丁質產品在食品、農業和醫藥等領域的應用。目前,幾丁質在食品工業中主要被用作膜材料和抗菌劑等。
幾丁質降解的方法目前主要有3 種:化學法、物理法和酶法。化學法通常是使用強酸(如鹽酸)使幾丁質的糖苷鍵斷裂,該方法不僅反應重復性差,而且會浪費大量的化學試劑,同時也會對環境造成污染。物理法主要是采用機械力、微波和超聲等降解幾丁質,目前國內外研究較少,一般不作為降解的主要方法,主要是用于輔助其他方法來共同降解幾丁質。相對于其他方法,酶法能夠在溫和的條件下降解幾丁質,并且降解過程以及降解產物都可控制,是目前較為理想的幾丁質降解方法。幾丁寡糖是幾丁質的降解產物,其分子質量低、水溶性好、易于分散和吸收。目前,幾丁寡糖的工業化制備仍然非常復雜,且成本較高。酶解反應條件溫和,且對環境友好,因此,酶法降解幾丁質為制備幾丁寡糖提供了一條可行的途徑。目前,幾丁質酶難以獲取而且價格昂貴,因此,一些非特異性酶被用來代替幾丁質酶降解幾丁質。這些非特異性酶種類多、來源廣泛、價格低廉,甚至有些非特異性酶對幾丁質的降解效率高于幾丁質酶。
幾丁質分子質量高,分子內和分子間氫鍵強,結晶度高,不溶于水、稀酸、堿性溶液和一般的有機溶劑,導致其難以酶促降解。因此,在酶解前可以對幾丁質進行改性預處理。目前,已有多種物理方法改性幾丁質的報道,包括超微粉碎、蒸汽爆破、瞬時彈射蒸汽爆破、超聲和高壓均質等。此外,也有使用有機溶劑、離子液體和超臨界水對幾丁質進行化學預處理的報道。蒸汽爆破和瞬時彈射蒸汽爆破原理相似,利用蒸汽強大的滲透力破壞幾丁質的晶體結構,但該方法對幾丁質的乙酰化會造成極大的破壞。超聲能夠打破幾丁質相對較弱的氫鍵和范德華力,但是,經過凍干后樣品會恢復晶體結構,結晶度不會降低,甚至會稍微升高。有機溶劑本身就具有毒性,并且會對環境造成一定的污染。離子液體價格較為昂貴。超臨界水對處理條件要求較高。而超微粉碎和高壓均質可以滿足幾丁質對溶劑的需要,破壞幾丁質的結晶結構,削弱分子間的氫鍵網絡,并且操作簡單、成本較低。然而目前鮮見關于超微粉碎和高壓均質聯合處理幾丁質的報道。
因此,為了打破幾丁質的分子內和分子間的氫鍵網絡,降低其分子質量,破壞其晶體結構,提高其酶促降解效率,本研究采用超微粉碎和高壓均質兩種物理方法聯合處理幾丁質;測定未處理和處理后的幾丁質平均粒徑、比表面積、孔隙體積、黏均分子質量、體積密度、振實密度和膨脹比率等理化特性;同時,采用傅里葉變換紅外(Fourier transform infrared,FTIR)光譜法、元素分析、X射線衍射(X-ray diffraction,XRD)、熱重-差示掃描量熱(thermogravimetric-differential scanning calorimetry,TG-DSC)法、掃描電子顯微鏡(scanning electron microscopy,SEM)對其微觀結構進行表征;最后,使用木瓜蛋白酶和纖維素酶對幾丁質進行降解,探究超微粉碎和高壓均質聯合處理對幾丁質酶解反應是否起到促進作用。
1 材料與方法
1.1 材料與試劑
幾丁質 生工生物工程(上海)股份有限公司。
無水LiCl 上海源葉生物科技有限公司;磷酸鹽緩沖液(phosphate buffered saline,PBS)、木瓜蛋白酶(20萬 U/g) 北京博奧拓達科技有限公司;,-二甲基乙酰胺 福晨(天津)化學試劑有限公司;纖維素酶(3 U/mg) 北京索萊寶科技有限公司;KBr 國藥集團化學試劑有限公司。
1.2 儀器與設備
電子天平 上海越平科學儀器有限公司;DE-100g萬能粉碎機 浙江紅景天工貿有限公司;SHA-C水浴恒溫振蕩器、79-2雙向磁力攪拌器 常州國宇儀器制品有限公司;QS-100S多功能高效球磨儀 五洲鼎創(北京)科技有限公司;APV-1000高壓均質機 德國APV公司;LGJ-12型冷凍干燥機 北京四環起航科技有限公司;721G可見分光光度計 上海儀電分析儀器有限公司;Mastersizer 2000激光粒度儀、Zetasizer Nano ZS90納米粒度及Zeta電位分析儀 英國馬爾文帕納科公司;NOVA-2000e比表面積及孔徑測試分析儀 美國康塔儀器公司;烏氏黏度計 上海申宜玻璃制品有限公司;IRAffinity-1S傅里葉變換紅外光譜儀 日本島津公司;EuroEA元素分析儀 意大利歐維特公司;Ultima IV X射線衍射儀 日本理學公司;STA 449F3同步熱分析儀德國耐馳儀器制造有限公司;MERLIN Compact掃描電子顯微鏡 德國蔡司公司。
1.3 方法
1.3.1 幾丁質的改性處理
將商品化幾丁質記作原幾丁質(raw chitin,RC),分別進行超微粉碎(ultra-micro grinding,UMG)、高壓均質(high-pressure homogenization,HPH)和超微粉碎-高壓均質聯合處理(ultra-micro grinding-high-pressure homogenization,UMG-HPH)。
1.3.1.1 幾丁質的超微粉碎處理
使用球磨儀對RC進行UMG處理。分別將2 g RC裝入球磨儀兩個腔室中,轉速設置為1 400 r/min,時間設置為30 min,得到超微粉碎幾丁質(ultra-micro grinding chitin,UMGC)。研磨前將腔室、研磨球和樣品于液氮中浸泡5 min。
1.3.1.2 幾丁質的高壓均質處理
將RC配制成懸濁液(30 g/L),然后進行HPH處理,壓力為40 MPa,時間為10 min,收集均質后的懸濁液,真空冷凍干燥,得到高壓均質幾丁質(high-pressure homogenization chitin,HPHC)。
1.3.1.3 幾丁質的超微粉碎和高壓均質聯合處理
按照1.3.1.1節方法將RC制備成UMGC,然后用蒸餾水將UMGC配制成懸濁液(30 g/L),再按照1.3.1.2節方法對其進行HPH處理,收集均質后的懸濁液,真空冷凍干燥,得到超微粉碎-高壓均質幾丁質(ultra-micro grinding-high-pressure homogenization chitin,UMG-HPHC)。
1.3.2 理化性質的測定
1.3.2.1 粒徑的測定
參考史早等的方法,RC的粒徑分布采用激光粒度儀測定,UMGC、HPHC和UMG-HPHC的粒徑分布采用納米粒度及Zeta電位分析儀測定,均在蒸餾水折射率1.33的條件下進行。取0.5 g樣品于蒸餾水中充分振蕩混勻,用膠頭滴管吸取上述樣品并緩慢加入裝有500 mL水的燒杯中,同時進行攪拌,直到儀器顯示的遮光度在測定范圍內緩慢波動時,停止滴加樣品并記錄儀器數據。
1.3.2.2 比表面積和孔隙體積的測定
參考Tian Zhiqing等的方法,使用比表面積及孔徑測試分析儀測定。使用N吸附/脫附等溫線法測定幾丁質的比表面積和孔隙體積。測定前,幾丁質在100 ℃脫氣12 h。
1.3.2.3 特性黏度和黏均分子質量的測定
參考張建旭等的方法測定幾丁質的特性黏度[],溶劑為質量分數5%的LiCl/,-二甲基乙酰胺溶液。[]的計算如式(1)所示。

式中:為/,為樣品溶液流出烏氏黏度計所用時間/s,為溶劑流出烏氏黏度計所用時間/s;為樣品溶液質量濃度/(g/mL)。
黏均分子質量(viscosity-average molecular mass,)通過Mark-Houwink-Sakurada方程(式(2))確定。

式中:為擴張因子(0.69);為比例系數(0.24 cm/g)。
1.3.2.4 體積密度、振實密度和膨脹比的測定
參考Tan等的方法測定體積密度、振實密度和膨脹比。
體積密度:稱量容積為100 cm的空杯質量(/g),將幾丁質樣品自由填充到杯中,去掉多余粉末,使粉末與杯頂部齊平,然后稱質量(/g)。體積密度按公式(3)計算。
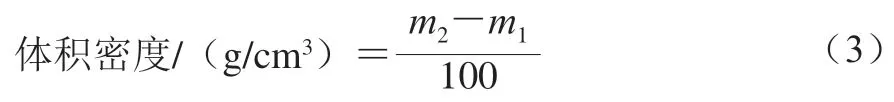
振實密度:稱量容積為100 cm的空杯質量(/g),將幾丁質樣品自由填充到杯中,敲擊100 次振實,去掉多余粉末,使粉末與杯頂部齊平,然后稱質量(/g)。振實密度按公式(4)計算。
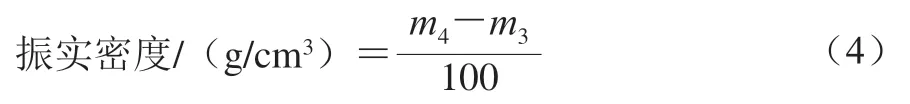
膨脹比:定義為未處理幾丁質的體積密度(或振實密度)與處理后幾丁質的體積密度(或振實密度)的比值。
1.3.3 微觀結構的表征
1.3.3.1 傅里葉變換紅外光譜分析
參考Delezuk等的方法,采用壓片法進行FTIR分析。取200 mg溴化鉀與2 mg幾丁質樣品于瑪瑙研缽中充分研磨,壓制成圓片,在FTIR儀上進行掃描,掃描范圍為4 000~400 cm,分辨率為4 cm,掃描次數32。
1.3.3.2 脫乙酰度的測定
參考Zhang Wenchang等的方法,采用元素分析儀測定幾丁質樣品中C和N的相對含量,脫乙酰度(deacetylation degree,DD)按式(5)計算。
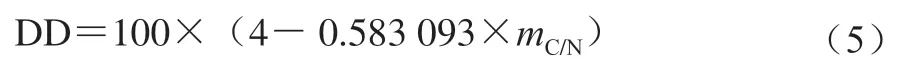
式中:是幾丁質樣品中C和N的質量比。
1.3.3.3 X射線衍射分析
參考張建旭等的方法,使用X射線衍射儀(靶:Cu-Kα;波長:1.541 8 nm;電壓:40 kV;電流:40 mA)對幾丁質樣品進行分析,掃描速率為10°/min,掃描范圍為5°~60°。(110)晶面和(020)晶面結晶度指數(crystallinity index,CrI)分別按公式(6)、(7)計算。
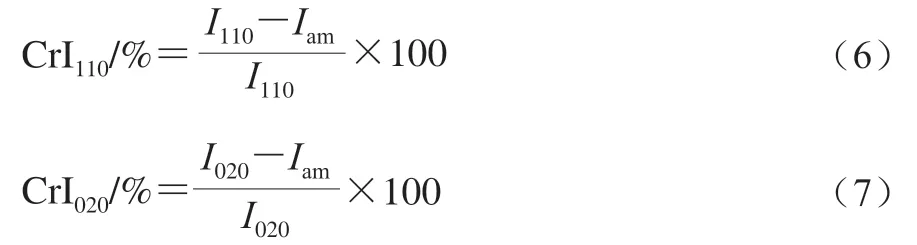
式中:和分別是2=22°和2=10°處的最大衍射強度;為2=16°處的非晶衍射強度。
1.3.3.4 熱重-差示掃描量熱綜合同步分析
參考Zhang Wenchang等的方法,使用同步熱分析儀對幾丁質進行TG-DSC分析。在N氣氛下,升溫范圍為30~600 ℃,升溫速率為10 ℃/min,收集升溫過程中的數據。
1.3.3.5 掃描電子顯微鏡觀察
參考Tian Zhiqing等的方法,將干燥的幾丁質樣品均勻地固定在SEM進樣臺上,真空條件下離子濺射噴金,采用SEM放大5 000 倍和50 000 倍進行微觀結構觀察。
1.3.4 幾丁質的酶解反應
1.3.4.1 木瓜蛋白酶對幾丁質的酶解反應
參考杜敬河的方法,將幾丁質樣品加入PBS(0.01 mol/L、pH 7.2)中配制底物溶液(50 mg/mL)。取1 mL底物溶液,加入1 mL木瓜蛋白酶溶液(10 mg/mL),于水浴搖床(37 ℃、180 r/min)中反應210 min。反應結束后,煮沸20 min,12 000 r/min離心2 min。同時設置對照組(向1 mL底物溶液中加入1 mL預先滅活的木瓜蛋白酶溶液,其余處理同上)。最后,使用二硝基水楊酸法測定上清液中還原糖含量。
1.3.4.2 纖維素酶對幾丁質的酶解反應
參考杜敬河的方法,將幾丁質樣品加入醋酸-醋酸鈉緩沖溶液(1 mol/L、pH 4.8)中,配制成底物溶液(50 mg/mL)。取1 mL底物溶液,加入1 mL纖維素酶溶液(10 mg/mL),于水浴搖床(40 ℃、180 r/min)中反應210 min,反應結束后,煮沸20 min,12 000 r/min離心2 min。同時設置對照組(向1 mL底物溶液中加入1 mL預先滅活的纖維素酶溶液,其余處理同上)。使用二硝基水楊酸法測定上清液中還原糖含量。
1.4 數據處理與分析
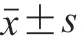
2 結果與分析
2.1 UMG和HPH聯合處理對幾丁質理化性質的影響
2.1.1 幾丁質樣品的外觀變化
如圖1所示,RC經過UMG處理后,UMGC粉末明顯細化,并且出現了二次團聚現象;再經過HPH處理,所得UMG-HPHC粉末的質地明顯蓬松,并且出現了更多的孔隙。

圖1 RC(A)、UMGC(B)和UMG-HPHC(C)的外觀圖像Fig. 1 Appearance images of raw chitin (RC) (A), ultra-micro grinding chitin (UMGC) (B) and ultra-micro grinding-high-pressure homogenization (UMG-HPHC) (C)
2.1.2 幾丁質樣品的粒徑變化
由表1可知,UMG和HPH處理會顯著降低幾丁質的粒徑(<0.05)。與RC相比,經UMG、HPH以及UMG-HPH處理后,、、和平均粒徑均顯著減小,其中,UMG-HPHC較RC分別減小99.31%、99.72%、99.82%和99.71%。這表明UMG的連續擠壓和沖擊力作用以及HPH的高速剪切和對流碰撞作用使幾丁質顆粒得到細化,并且兩種方法聯合處理后粉末細化程度更高。

表1 RC、UMGC、HPHC和UMG-HPHC的粒徑分布Table 1 Particle size distribution of RC, UMGC, HPHC and UMG-HPHC
2.1.3 幾丁質樣品的比表面積和孔隙體積變化
由表2可知,UMGC、HPHC和UMG-HPHC的比表面積分別較RC增加-37.88%、209.23%和85.11%,孔隙體積分別增大-38.71%、177.42%和29.03%。在球磨儀中進行UMG處理之初,幾丁質粒度細化,改變了幾丁質的表面結構,使幾丁質的表面結構變得更致密;隨著UMG處理的進行,幾丁質的細小顆粒又發生了二次團聚,最終導致比表面積減小。這一現象與Zhang Wenchang等的觀察結果一致。HPH的空化效應能夠增大幾丁質比表面積和孔隙體積,這使得幾丁質的作用面積增大,提高了幾丁質的應用潛力。
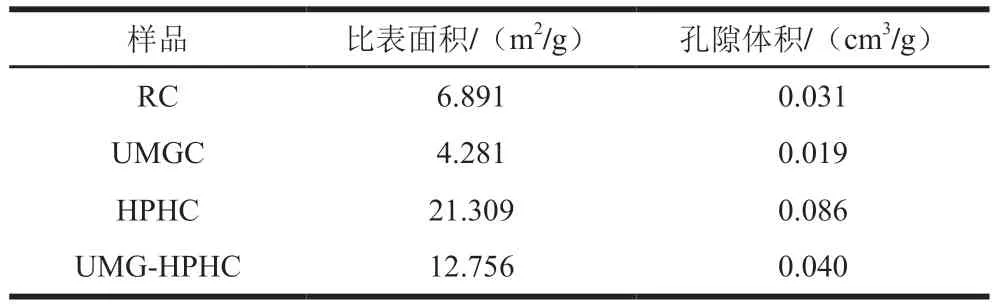
表2 RC、UMGC、HPHC和UMG-HPHC的比表面積和孔隙體積Table 2 Specific surface areas and pore volumes of RC, UMGC,HPHC and UMG-HPHC
2.1.4 幾丁質樣品的黏均分子質量變化
幾丁質的生物學效應和力學性能高度依賴于其分子質量。因此,幾丁質的是一個非常重要的性能評價參數。UMG和HPH均為機械處理,會導致分子鏈的斷裂和分子質量降低。分子質量的降低是由于糖苷鍵的斷裂,糖苷鍵斷裂后產生的自由基會進一步誘導其他活性基團的形成,從而有利于幾丁質進一步降解,提高其溶解性。
由表3可知,經UMG、HPH以及UMG-HPH處理后,樣品的特性黏度和均發生了不同程度的降低。與RC相比,UMGC、HPHC和UMG-HPHC的分別降低了75.32%、80.05%和85.80%。由此可以看出,UMG和HPH處理都能使幾丁質的降低,兩種方法聯合可以得到更低的幾丁質。

表3 RC、UMGC、HPHC和UMG-HPHC的特性黏度和黏均分子質量Table 3 Intrinsic viscosity and viscosity-average molecular masses of RC, UMGC, HPHC and UMG-HPHC
2.1.5 幾丁質樣品的體積密度、振實密度及膨脹比變化
由表1、4可知,RC經過UMG處理后平均粒徑大幅度降低,導致膨脹比下降。HPHC和UMG-HPHC分別是由RC和UMGC經HPH處理得到的,經振實密度計算的膨脹比較處理前分別增大3.98 倍和1.02 倍,說明HPH能夠使幾丁質的質地更加蓬松,表明幾丁質的孔隙體積變大,這與比表面積及孔隙體積分析結果相印證。
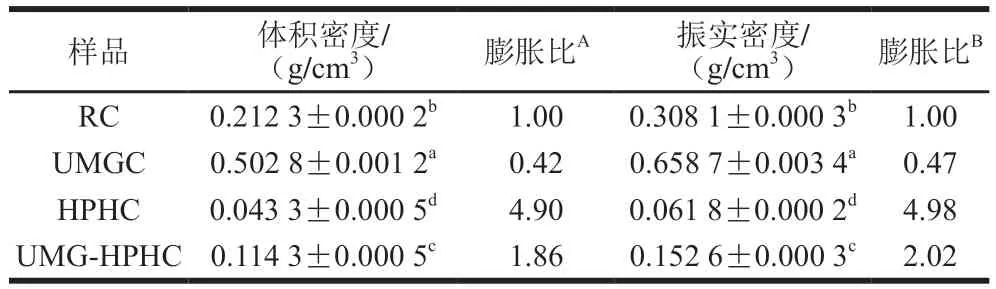
表4 RC、UMGC、HPHC和UMG-HPHC的體積密度、振實密度和膨脹比Table 4 Bulk densities, tap densities and expansion ratios of RC,UMGC, HPHC and UMG-HPHC
2.2 UMG和HPH聯合處理對幾丁質微觀結構的影響
2.2.1 FTIR分析結果
紅外光譜中吸收峰的位置及強度的變化與原子振動頻率及其化學組成、化學鍵類型有著密切的關系。由圖2可知,UMGC、HPHC和UMG-HPHC的峰位置和形狀基本與RC一致,說明UMG和HPH處理對幾丁質的化學結構沒有明顯影響。在3 448 cm附近的吸收峰由O—H伸縮振動引起,3 263 cm附近的吸收峰由N—H伸縮振動引起,這兩個吸收峰受氫鍵的影響較大。UMGC、HPHC和UMG-HPHC與RC相比,在3 448 cm和3 263 cm附近吸收峰強度提高,表明UMG和HPH處理破壞了幾丁質之間氫鍵網絡,使更多的羥基基團暴露。另外,2 961~2 840 cm處的吸收峰屬于C—H伸縮振動。以上幾種吸收峰均是多糖的特征吸收峰,表明樣品的主要成分是含氮糖類物質。1 662 cm和1 631 cm處的兩個吸收峰是-幾丁質在酰胺I帶(C=O)的特征吸收峰,而1 564 cm和1 315 cm處的吸收峰分別代表酰胺II帶(N—H)和酰胺III帶(C—N)。1 160~1 020 cm為C—O伸縮振動的吸收峰,896 cm附近的吸收峰是由幾丁質的-糖苷鍵伸縮振動引起的,896 cm處的吸收峰強度變弱,表明UMG和HPH處理可能使幾丁質中的部分糖苷鍵斷裂,這與黏均分子質量分析結果相印證。
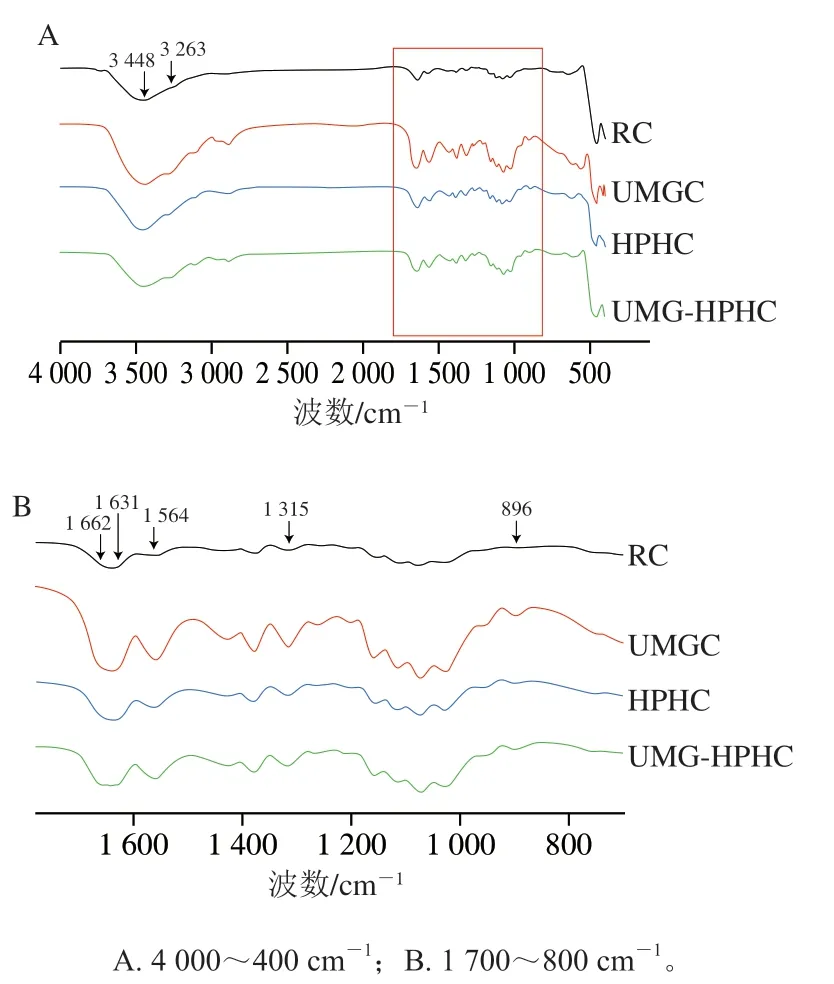
圖2 RC、UMGC、HPHC和UMG-HPHC的FTIR光譜Fig. 2 FTIR spectra of RC, UMGC, HPHC and UMG-HPHC
2.2.2 脫乙酰度測定結果
由表5可知,通過UMG處理,RC的DD從7.84%下降到6.14%,再繼續HPH處理,DD下降到5.84%。而通過HPH單一處理,RC的DD從7.84%下降到3.24%。這些結果表明,UMG和HPH均不能增大幾丁質的DD。完全乙酰化幾丁質的理論N相對含量為6.9%,本研究中不同幾丁質樣品的N相對含量在6.08%~6.48%之間,與之對應,UMGC、HPHC和UMG-HPHC的DD均有所降低,尤其是HPH單一處理的樣品DD降幅最大。目前,高乙酰度的幾丁寡糖由于在醫藥和農業應用中能夠改善生物反應而備受關注。乙酰基在結晶度較低、比表面積較大的改性幾丁質中的保留將有利于乙酰化寡糖的合成。
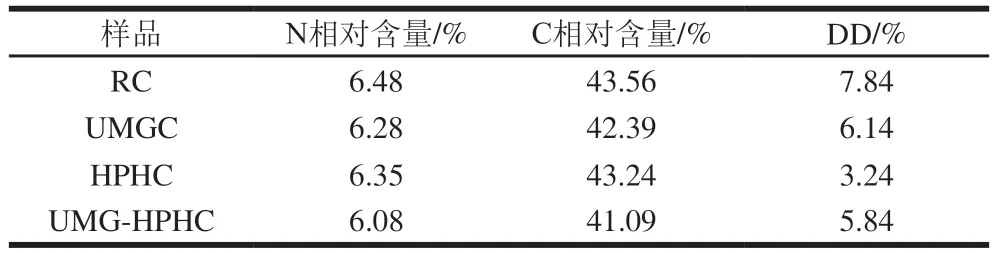
表5 RC、UMGC、HPHC和UMG-HPHC的脫乙酰度分析Table 5 Deacetylation degree analysis of RC, UMGC, HPHC and UMG-HPHC
2.2.3 XRD分析結果
由圖3可見,2在19.2°附近為幾丁質的主衍射峰,同時在9.4°、23.4°和26.3°附近有較小的衍射弱峰,呈現出典型的幾丁質晶體結構。與RC相比,UMGC、HPHC和UMG-HPHC的衍射峰位置沒有明顯變化,說明幾丁質的晶型沒有發生改變。但是,衍射峰強度均有所降低,說明在處理過程中幾丁質結晶結構被破壞。結晶度與分子內和分子間氫鍵有著緊密關系,結晶度降低表明幾丁質的氫鍵網絡被破壞,這與FTIR光譜分析結果一致。在(110)晶面,UMGC、HPHC和UMG-HPHC的結晶度指數分別降低了12.68%、3.85%和17.49%,在(020)晶面,UMGC、HPHC和UMG-HPHC的結晶度指數分別降低了17.17%、4.08%和1.31%,表明經過UMG和HPH聯合處理后,UMG-HPHC的有序度和結晶度降低。
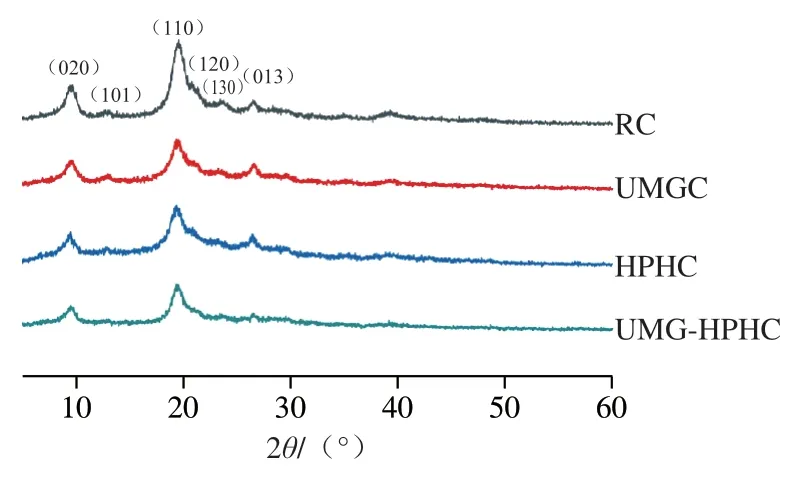
圖3 RC、UMGC、HPHC和UMG-HPHC的XRD圖Fig. 3 X-ray diffraction diagrams of RC, UMGC, HPHC and UMG-HPHC
2.2.4 TG-DSC綜合分析結果
由圖4A可見,幾丁質的熱分解過程主要分為3 個階段。第一個熱分解階段發生在40~140 ℃,該階段的質量損失是由幾丁質分子內部的自由水和結合水逐漸蒸發引起,HPHC和UMG-HPHC蒸發速率明顯高于RC,是由于結晶結構陰礙水的進出,低結晶度幾丁質的蒸發速率更快,蒸發溫度相對降低。
第二個質量損失階段發生在200~450 ℃,這是樣品質量損失率最高的階段,主要原因是幾丁質的分解。由于經過UMG處理的幾丁質粒徑減小,經過HPH處理的幾丁質比表面積增加,孔隙體積擴大,所以UMGC、HPHC和UMG-HPHC受熱面積增大,受熱更加均勻,使其分解速率加快。450 ℃之后,幾丁質分解進入平緩的第三個階段,此階段主要是熱解殘余物緩慢分解產生碳和灰分。
由圖4B可見,由于幾丁質的自由水和結合水的蒸發,所有樣品均在150 ℃之前有較寬的吸熱峰。4 個樣品的峰值不同,可能與結晶度有關。UMG-HPHC的峰值出現在94.14 ℃,較其他樣品峰值延遲,是因為結晶度的降低使幾丁質的吸水能力增強。
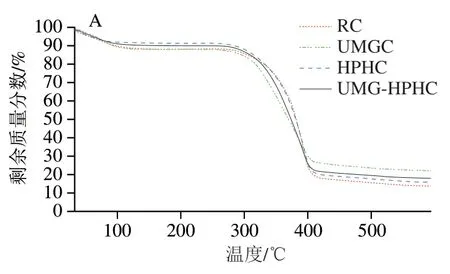
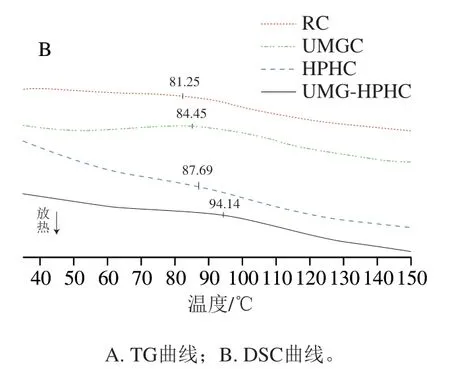
圖4 RC、UMGC、HPHC和UMG-HPHC的TG-DSC綜合分析Fig. 4 TG-DSC analysis of RC, UMGC, HPHC and UMG-HPHC
2.2.5 掃描電子顯微鏡觀察結果
由圖5可見,未經過處理的RC表面光滑、結構有序、孔洞分布均勻(圖5A、A);經UMG處理后的UMGC結構被破壞,表面呈現堆疊現象,孔洞消失(圖5B、B),并且可以觀察到幾丁質團聚形成聚集體,當幾丁質顆粒足夠小時,其粗糙度和顆粒表面的凹凸度等發生改變,都可能引起幾丁質發生聚集,這也是比表面積減小的原因;經HPH處理后的HPHC結構遭到嚴重破壞,形成雜亂的網狀多孔結構,并且能夠清晰地看到疏松多孔的蓬松結構(圖5C、C);UMG-HPHC的微觀結構(圖5D、D)和HPHC相似,具有疏松多孔的蓬松結構。以上結果表明,UMG和HPH處理對幾丁質的結構有明顯影響。
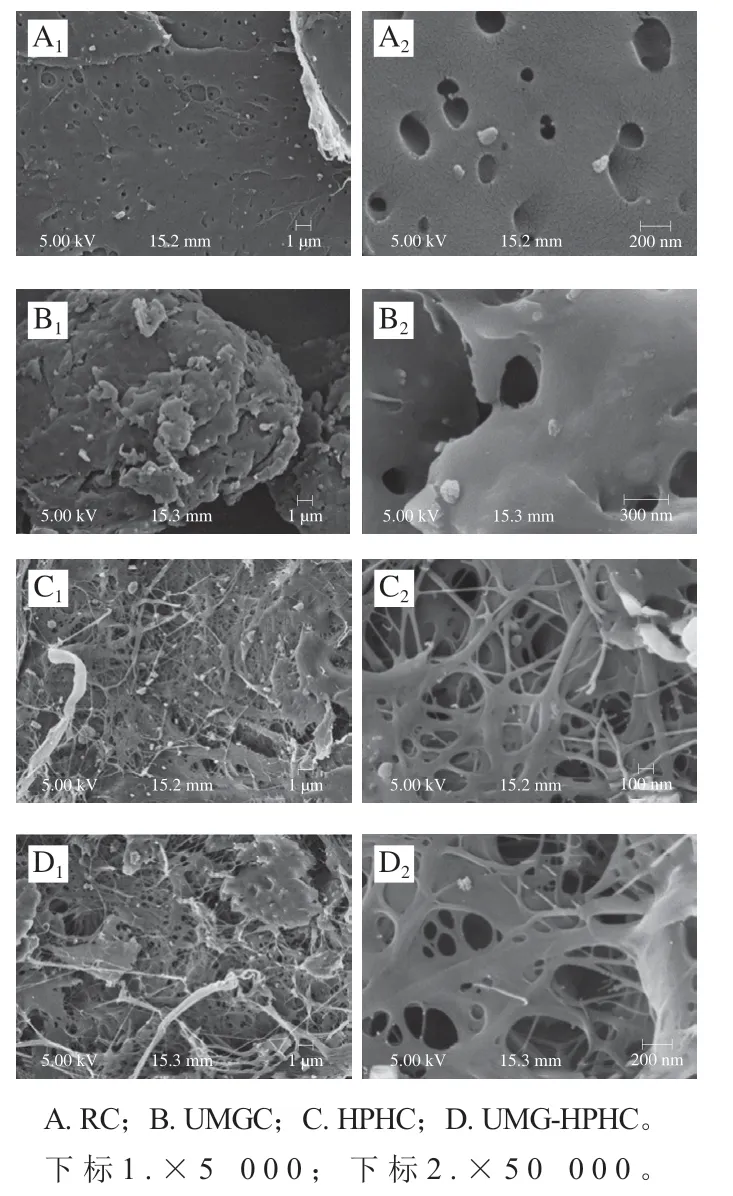
圖5 RC、UMGC、HPHC和UMG-HPHC的SEM圖Fig. 5 SEM images of RC, UMGC, HPHC and UMG-HPHC
2.3 UMG和HPH聯合處理對幾丁質的酶解效率的影響
如圖6所示,木瓜蛋白酶和纖維素酶降解4 種樣品的還原糖產量均為UMG-HPHC>HPHC>UMGC>RC。在木瓜蛋白酶和纖維素酶對幾丁質的降解中,UMG-HPHC的還原糖產量比RC分別提高了6.05 mg/mL和0.87 mg/mL,表明UMG和HPH處理對木瓜蛋白酶和纖維素酶降解幾丁質有積極作用,并且UMG和HPH聯合處理效果最佳,體現出一定的協同增效作用。經過聯合處理的改性幾丁質降低、結晶度減小、比表面積增大,形成網狀多孔結構。這些理化性質和微觀結構的變化均有助于改善幾丁質的溶解度,并可能增加酶與底物的結合位點,從而提高酶對幾丁質的降解效果。
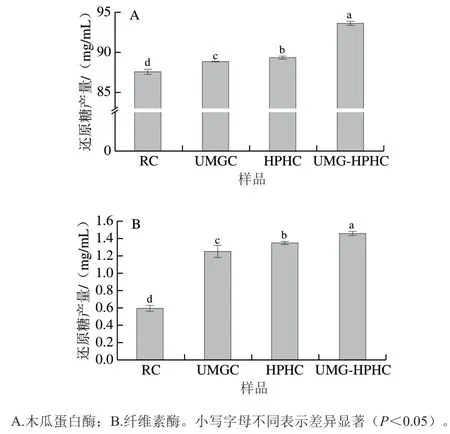
圖6 不同改性處理對幾丁質酶解效率的影響Fig. 6 Effects of different modification treatments on enzymatic degradation efficiency of chitin
3 結 論
為了打破幾丁質分子內和分子間的氫鍵網絡,降低其分子質量,破壞其晶體結構,以提高幾丁質的酶解效率,本實驗研究了UMG和HPH聯合處理對幾丁質理化特性和微觀結構的影響。結果表明,經過UMG單一處理后,幾丁質顆粒得到細化,結晶度降低。UMG和HPH聯合處理能夠得到既細化又蓬松的改性幾丁質粉末,并且改性后的幾丁質孔隙體積和比表面積增大,平均粒徑和降低,分子間氫鍵網絡被破壞,更多的羥基基團暴露,部分糖苷鍵斷裂,結晶度和熱穩定性降低,同時形成了網狀多孔結構,使更多的結合位點暴露。與RC和單一改性處理的幾丁質相比,聯合處理改性的幾丁質更易被木瓜蛋白酶和纖維素酶降解。此外,改性后幾丁質的脫乙酰度有所降低,為制備高乙酰度的幾丁寡糖提供了有利條件,也保證了幾丁質進一步衍生化的可能性和多樣性,如制備可溶性或具有功能特性的幾丁質衍生物等。綜上,UMG和HPH聯合處理有效改善了幾丁質的理化和結構特性,作為一種操作簡單且對環境友好的處理方式,可作為促進幾丁質酶促降解的一條有效途徑,同時提高幾丁質的應用潛力。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
兒童故事畫報(2019年5期)2019-05-26 14:26:14
產品可靠性報告(2017年7期)2017-09-05 09:49:12
Coco薇(2016年2期)2016-03-22 02:42:52
汽車觀察(2016年3期)2016-02-28 13:16:26
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
