一測多評法同時測定外用金黃散提取物中7種成分的含量
2022-10-14 05:08:48肖新玉楊小燕施文婷張蘭蘭
廣東藥科大學學報 2022年5期
肖新玉,楊小燕,施文婷,張蘭蘭
(1.佛山市南海區人民醫院,廣東 佛山 528225;2.廣東一方制藥有限公司,廣東 佛山 528225)
外用金黃散是佛山市南海區人民醫院根據中醫藥理論為基礎,由多年臨床經驗總結而成的有效方劑,基礎方源于明代陳實功所著《外科正宗》,由大黃、黃柏、姜黃、白芷、天花粉、生天南星、蒼術、厚樸、陳皮、甘草十味藥組成,具有清熱燥濕、活血化瘀、消腫止痛、收濕斂瘡的功效,臨床上主要用于痛風性關節炎、急性乳腺炎、急性闌尾炎等的治療[1-2]。外用金黃散在原方基礎上增加了乳香、冰片等四味藥材,增強了活血化瘀、消腫生肌的功效。目前,關于金黃散的研究較多集中于提取工藝和藥理作用方面[3-6];2020年版《中國藥典》規定如意金黃散的定量指標為姜黃素、小檗堿、蘆薈大黃素、大黃酸、大黃素、大黃酚、和大黃素甲醚7種,采用3種色譜方法,較為復雜[7];姚遠等[8]采用超高效液相色譜-質譜聯用(UPLC-MS)法同時測定如意金黃散中12種成分,所需對照品種類較多,檢測成本較高。
一測多評法(quantitative analysis of multicomponents by single-marker,QAMS)借助相對校正因子,只需測定一個成分(內標物),可實現對多個有效成分的定量分析,在提高檢測效率的同時降低了檢測成本,已在中藥質量控制方面得到廣泛應用[9]。QAMS法在建立過程中,需要通過測定內標物與其他待測成分的峰面積和濃度,計算內標物與其他待測成分的相對校正因子,相對校正因子是實現QAMS法的關鍵參數,所以在建立該方法時相對校正因子的取值標準選擇尤為重要。此外,計算值與外標法實測值之間的相似程度還與選定的內標物與其他待測成分間化學結構和理化性質的差異關系密切[10]。本研究通過建立一測多評法,以蘆薈大黃素為內標物,同時測定外用金黃散提取物中甘草苷、橙皮苷、甘草酸、大黃酸、歐前胡素和大黃素甲醚的含量,以期為外用金黃散提取物的質量控制及后續研究提供參考。
1 儀器與試藥
1.1 儀器
Thermo U3000型高效液相色譜儀(美國賽默飛公司);Waters Arc型高效液相色譜儀(美國沃特世公司);Milli-Q Direct型超純水系統(德國默克公司);XP26型百萬分之一天平、ME204E型萬分之一天平(瑞士梅特勒-托利多儀器有限公司);KQ-500DE型數控超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 試藥
甘草苷(批號:111610-201908,純度為95.0%)、橙皮苷(批號:110721-202019,純度為95.3%)、甘草酸銨(批號:110731-202021,純度為96.2%)、蘆薈大黃素(批號:110795-201710,純度為98.3%)、大黃酸(批號:110757-201607,純度為99.3%)、歐前胡素(批號:110826-201918,純度為99.0%)、大黃素甲醚(批號:110758-201817,純度為99.2%)對照品均購自中國食品藥品檢定研究院;乙腈、甲醇為色譜純(德國默克公司);磷酸為色譜純(天津市科密歐化學試劑有限公司);其他試劑為分析純。9批外用金黃散提取物(編號:S1~S9)由廣東一方制藥有限公司制備。
2 方法與結果
2.1 色譜條件[11]
色譜柱:Waters Xbridge C18柱(250 mm×4.6 mm,5.0 μm);流動相:乙腈(A)-0.1%磷酸溶液(B)梯度洗脫(0~16 min,19%~24%A;16~20 min,24%~30%A;20~30 min,30%~46%A;30~42 min,46%~55%A;42~60 min,55%~62%A;60~65 min,62%~19%A;65~70 min,19%A;);檢測波長:280 nm(0~20 min,甘草苷、橙皮苷)、254 nm(20.01~70 min,甘草酸、蘆薈大黃素、大黃酸、歐前胡素、大黃素甲醚);柱溫:30℃;進樣量:10 μL;流速:1.0 mL/min。
2.2 對照品溶液的制備
分別取甘草苷、橙皮苷、甘草酸銨、蘆薈大黃素、大黃酸、歐前胡素、大黃素甲醚對照品適量,精密稱定,加甲醇定容,制成質量濃度分別為92.598 6、77.334 8、65.154 6、72.999 8、85.880 0、64.184 6、66.844 8 μg/mL的混合對照品儲備液(甘草酸質量=甘草酸銨質量/1.0207)。
2.3 供試品溶液的制備
取外用金黃散提取物約0.4 g,精密稱定,置50 mL具塞錐形瓶中,精密加入甲醇25 mL,密塞,稱定質量,超聲處理(功率250 W,頻率40 kHz)30 min,取出,放冷后再稱定質量,用甲醇補足減失的質量,搖勻,濾過,取續濾液,即得。
2.4 方法學考察
2.4.1 專屬性試驗分別精密吸取對照品、供試品、陰性樣品溶液,按“2.1”項下色譜條件進樣測定,結果見圖1。可見,各成分色譜峰均具有良好的分離度,與對照品色譜峰保留時間一致,并且未在相應陰性樣品中檢測出,表明該方法專屬性良好。

圖1 專屬性試驗HPLC色譜圖Figure 1 HPLC chromatogram of specificity test
2.4.2 線性關系考察分別精密吸取“2.2”項下混合對照品溶液,加甲醇稀釋制成系列濃度的混合對照品溶液,按“2.1”項下色譜條件進樣測定,記錄甘草苷、橙皮苷、甘草酸、蘆薈大黃素、大黃酸、歐前胡素、大黃素甲醚的峰面積。以對照品溶液質量濃度為橫坐標(x),峰面積為縱坐標(y)進行線性回歸,結果見表1,表明各成分在各自質量濃度范圍內均呈良好的線性關系。

表1 線性關系考察結果Table 1 Results of linear relationship investigation
2.4.3 精密度試驗精密吸取“2.2”項下混合對照品溶液,按“2.1”項下色譜條件連續進樣6次,計算得各色譜峰峰面積的RSD均小于3%,表明儀器精密度良好。
2.4.4 重復性試驗取外用金黃散提取物(編號:S1),精密稱定,按“2.3”項下方法平行制備6份供試品溶液,按“2.1”項下色譜條件進樣測定,計算得各色譜峰峰面積的RSD均小于3%,表明方法重復性良好。
2.4.5 穩定性試驗取外用金黃散提取物(編號:S1),精密稱定,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件分別于制備0、2、4、8、12、24 h后進樣測定,計算得各色譜峰峰面積的RSD均小于3%,表明供試品溶液在24 h內穩定性良好。
2.4.6 加樣回收率試驗取同一批次已知成分含量的外用金黃散提取物共9份,分為3組,每份取約0.2 g,精密稱定后置于錐形瓶中,分別按低、中、高濃度精密加入混合對照品溶液適量,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積,計算加樣回收率及RSD,結果見表2。結果顯示,各成分的平均加樣回收率分別是:甘草苷101.6%(RSD=1.81%)、橙皮苷99.8%(RSD=2.44%)、甘草酸99.3%(RSD=1.90%)、蘆薈大黃素97.7%(RSD=2.42%)、大黃酸97.7%(RSD=2.91%)、歐前胡素101.0%(RSD=3.30%)和大黃素甲醚99.1%(RSD=1.60%),表明該方法準確度良好。
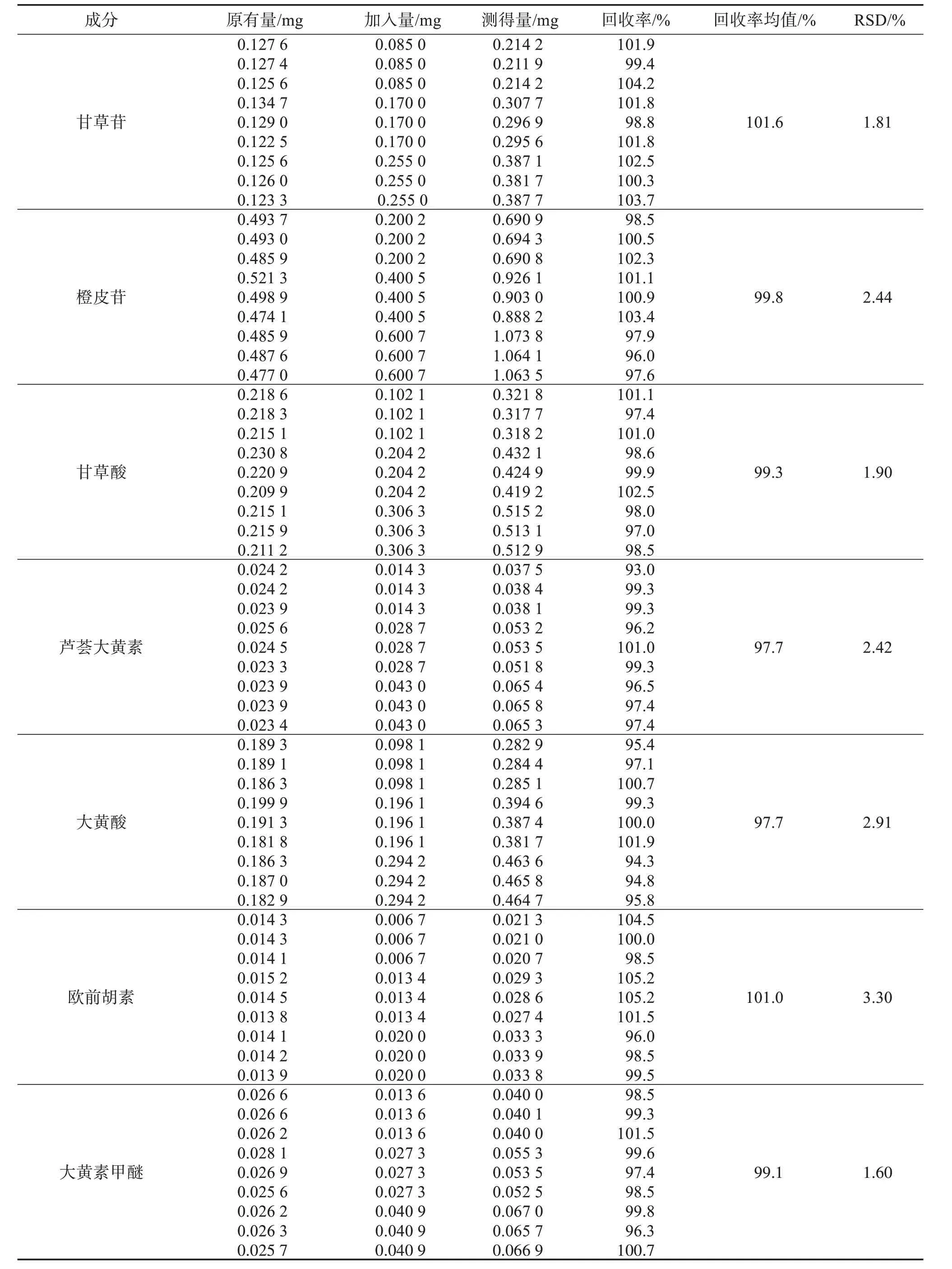
表2 外用金黃散提取物中7種成分的加樣回收率試驗結果Table 2 Test results of sample addition recovery rate of 7 components in Jinhuang powder for external use
2.5 相對校正因子的確定[12-13]
精密吸取“2.4.2”項下混合對照品溶液,按“2.1”項下色譜條件進樣測定,以蘆薈大黃素為內標物,計算其他6種成分的相對校正因子(fs/k),公式為fs/k=fs/fk=(As×Ck)/(Ak×Cs),其中As為內標物峰面積,Cs為內標物質量濃度,Ak為待測成分峰面積,Ck為待測成分質量濃度,結果見表3。

表3 不同濃度對照品溶液中各成分的相對校正因子Table 3 Relative correction factors of various constituents of different concentration of standard sample solution(n=7)
2.6 耐用性和系統適應性研究
2.6.1 不同色譜系統及色譜柱對相對校正因子的影響[14]以蘆薈大黃素為內標物,考察其他6種成分在Waters Arc型、Thermo U3000型2種高效液相色譜系統,以及Waters Xbridge C18(250 mm×4.6 μm,5 μm)、YMC Triart C18(250 mm×4.6 μm,5 μm)、Agilent ZORBAX Extend-C18(250 mm×4.6 μm,5 μm)3種色譜柱下的相對校正因子,結果見表4。可見,各成分的RSD均小于3%,表明不同色譜系統和色譜柱對各成分相對校正因子無顯著影響。

表4 不同色譜系統、色譜柱下各成分的相對校正因子Table 4 Effects of different instruments and columns on relative correction factors
2.6.2 不同柱溫對相對校正因子的影響[14]采用Thermo U3000型高效液相色譜儀、Waters Xbridge C18色譜柱,考察柱溫25、30、35℃對各成分相對校正因子的影響,結果見表5。可見,不同柱溫條件下各成分的RSD均小于3%,表明柱溫對各成分相對校正因子無顯著影響。
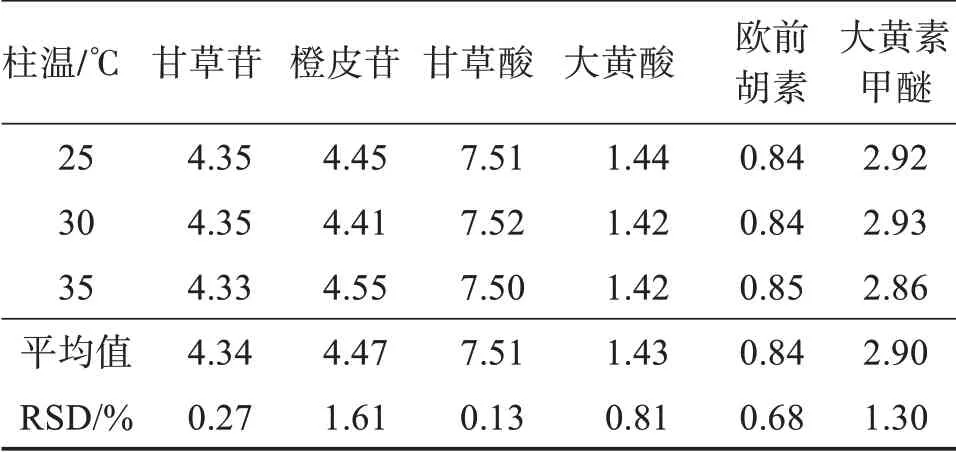
表5 不同柱溫下各成分的相對校正因子Table 5 Effects of different column temperatures on relative correction factors
2.6.3 不同流速對相對校正因子的影響[14]采用Thermo U3000型高效液相色譜儀、Waters Xbridge C18色譜柱,考察流速0.8、1.0、1.2 mL/min對各成分相對校正因子的影響,結果見表6。可見,不同流速條件下各成分的RSD均小于3%,表明不同流速對各成分相對校正因子無顯著影響。

表6 不同流速下各成分的相對校正因子Table 6 Effects of different flow rates on relative correction factors
2.6.4 不同進樣體積對相對校正因子的影響[14]采用Thermo U3000型高效液相色譜儀、Waters Xbridge C18色譜柱,考察進樣體積2、4、6、8、10、15 μL對各成分相對校正因子的影響,結果見表7。可見,各成分的RSD均小于3%,表明進樣體積的波動對各成分相對校正因子無顯著影響。

表7 不同進樣體積下各成分的相對校正因子Table 7 Effects of different injection volume on relative correction factors
2.7 待測成分色譜峰的定位[15]
在QAMS的應用中,目前常用的色譜峰定位方法有相對保留值法、保留時間差法、時間校正法、對照提取物法等。本研究采用相對保留時間進行待測組分色譜峰的定位,以蘆薈大黃素為內標物,考察其他6成分在Thermo U3000型和Waters Arc型2種高效液相色譜系統,以及3根不同廠家的色譜柱(Waters Xbridge C18、Agilent ZORBAX Extend-C18、YMC Triart C18)的相對保留時間,結果見表8。可見,各成分的RSD均小于3%,表明采用相對保留值法對待測成分的定位是可行的。

表8 不同色譜系統、色譜柱下各成分的相對保留時間Table 8 Effects of different instruments and columns on relative retention time
2.8 一測多評法與外標法測定結果比較
取9批外用金黃散提取物適量,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,分別采用外標法和一測多評法測定各成分質量分數,利用SPSS 20.0軟件對兩組檢測結果進行Pearson相關系數(r)分析,結果見表9。可見,2種方法的含量測定結果基本一致,相關系數r均為1.000。甘草苷、橙皮苷、甘草酸、大黃酸、歐前胡素、大黃素甲醚的相對標準偏差范圍分別為2.86%~3.60%、0.83%~1.04%、0.15~3.10%、0.74%~3.86%、0.23%~4.03%、0.02%~4.02%,均小于5.0%,表明2種方法計算結果無明顯差異。
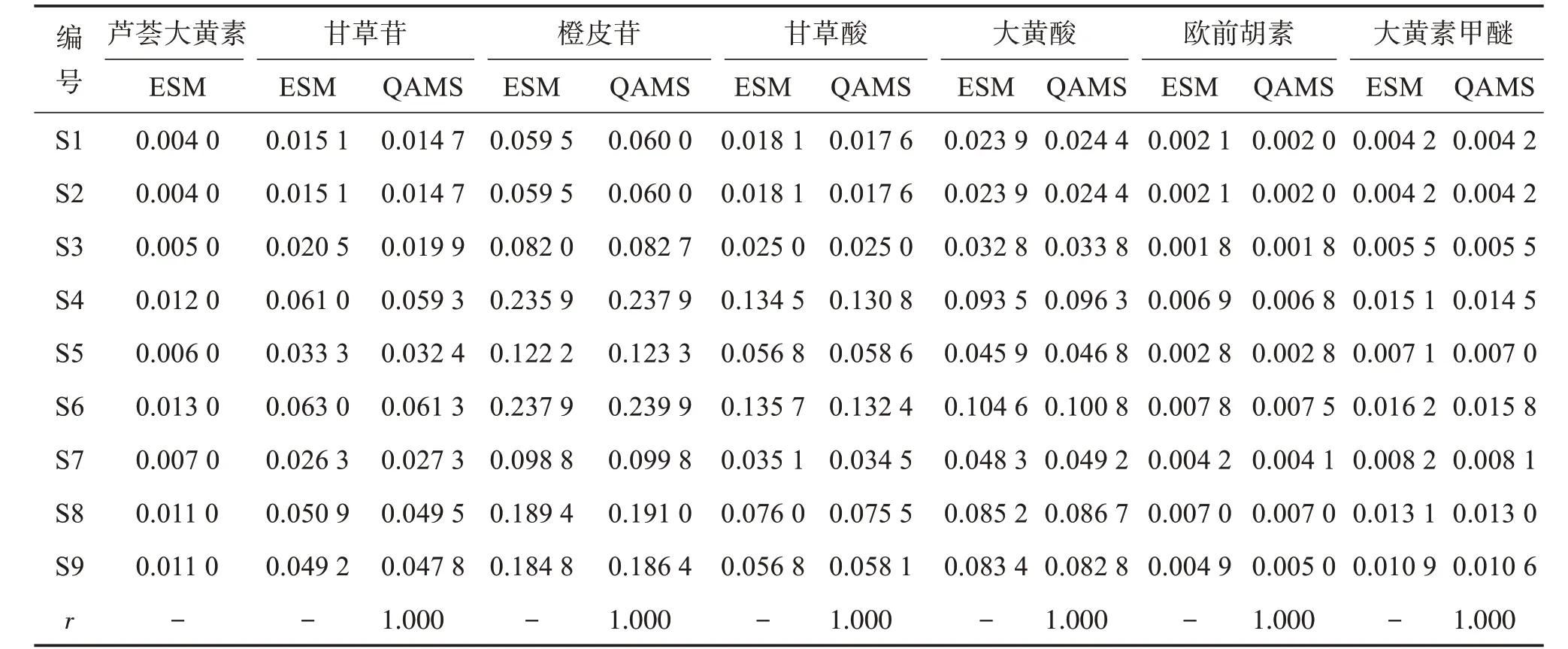
表9 外標法與一測多評法測定結果比較Table 9 Comparison of results obtained by external standard method and QAMS method(n=3) w/%
3 討論
目前,中藥化學標準品的數量和種類仍不能滿足質量檢測的需要,實際應用中常采用分別測定有效成分含量的方法來控制中藥及其制劑的質量,檢測效率較低,檢測成本偏高。一測多評法利用中藥中各成分間的相對校正因子同時測定多個有效成分,實現了在對照品缺乏的情況下進行中藥多成分的質量控制,顯著降低了檢測成本,提高了工作效率,對于中藥及其制劑的研究與開發具有重要意義[16]。
外用金黃散處方中君藥為苦寒解毒藥,如大黃、黃柏;臣藥為燥濕化痰行氣的天南星、陳皮、厚樸和破血行氣藥姜黃;使藥為辛溫祛風燥濕、消腫止痛藥,如白芷[17]。其藥理活性涉及到的化學成分有蘆薈大黃素、大黃酸、大黃素、大黃酚、大黃素甲醚、橙皮苷、甘草酸、歐前胡素等,這些化學成分可通過多種途徑發揮協同作用,因此,采用多指標進行藥品質量控制意義重大[18]。
本研究通過建立QAMS法同時測定外用金黃散提取物中甘草苷、橙皮苷、甘草酸、大黃酸、歐前胡素、大黃素甲醚的含量,該方法與外標法所測得的含量無顯著差異,說明建立的相對校正因子可靠可行,可達到1個對照品同時測定7個成分含量的目的,同時可降低多組分質量控制的檢測成本,可為外用金黃散提取物質量標準的建立提供參考。
