氣相色譜法測定魚油脂肪乳注射液中三酰甘油的含量
2022-10-14 05:07:56謝春燕曾少群岳峰
廣東藥科大學學報 2022年5期
謝春燕,曾少群,岳峰
(1.廣東嘉博制藥有限公司,廣東 清遠 511517;2.廣東靜脈脂肪乳工程中心,廣東 清遠 511517)
魚油在防治冠心病、高血壓、糖尿病、關節炎等疾病,以及自身免疫性病變和癌癥中有重要作用,具有極高的營養和臨床價值。魚油中的脂肪酸成分有EPA(二十二碳六烯酸)、DHA(二十碳五烯酸)、棕櫚酸(palmitic acid)、油酸(oleic acid)、9-十六碳烯酸(palmitoleicacid)、肉豆蔻酸(myristic acid)、花生四烯酸(arachidonic acid)、亞油酸(linoleic acid)、亞麻酸(alpha-linolenic acid),其中主要成分DHA和EPA是人體必需的不飽和脂肪酸[1-2]。
EPA和DHA主要有游離型、乙酯型和甘油酯型3種形式。研究結果顯示,EPA和DHA乙酯在人體中消化和吸收比較困難,可能存在安全隱患,游離型的EPA和DHA容易氧化生成對人體有害的物質,口感不好,直接食用難以被接受。甘油酯型性質穩定、不易氧化,而且口感好,易被人體消化吸收[3]。天然魚油一般以EPA和DHA甘油酯的形式存在,含量較低,目前廣泛應用于乳制品、保健食品和醫藥產品中的是經精制的富含EPA和DHA甘油酯的魚油。
德國貝朗(B.Braun)公司開發的魚油中/長鏈脂肪乳注射液(商品名力保魚優,Lipoplus),2004年在歐洲上市,2014年在國內上市。該產品采用中鏈甘油三酸酯、大豆油和ω-3精制魚油組合,具有炎癥調節作用及減少感染和并發癥的作用。當口服或腸內營養不足、有禁忌或無法進食時,本品作為腸外營養的組成部分,提供包括人體必需ω-6、ω-3脂肪酸在內的脂肪。目前國內已有兩個廠家仿制力保魚優的產品并獲得批準上市,還有費森尤斯卡比生產的含精制魚油的多種油脂肪乳注射液(C6~24)。含魚油的脂肪乳注射液在臨床上越來越受到重視,可為機體提供必需的能量和代謝底物。此外,還可通過抑制炎癥反應增加機體免疫力、改善防御能力、減輕氧化應激、維護胃腸功能與結構和促進機體康復,拓展了其治療臨床危重癥的新途徑[4-5]。
本文測定的魚油脂肪乳注射液參考力保魚優,以中鏈三酰甘油、大豆油和ω-3魚油為主藥。其中ω-3魚油為甘油酯型魚油,中鏈甘油三酸酯的主要成分為辛酸甘油酯和癸酸甘油酯,大豆油主要含油酸甘油酯、亞油酸甘油酯、亞麻酸甘油酯。本文研究的目的是同時測定魚油脂肪乳注射液的主要三酰甘油。直接測定三酰甘油的分析方法主要有高碘酸法、柱層析法、酶法、薄層色譜法(TLC)、氣相色譜法、高效液相色譜法等,其中化學法、柱層析法易出錯、操作步驟多、耗時長。酶法容易使甘油酯水解,導致脂肪酸位置鑒定困難。
目前儀器分析法是較為常用的方法[6],有氣相色譜法[7-8]、液相色譜法[9]、氣相色譜-質譜聯用法[10]、液相色譜-質譜聯用法[11]、紅外光譜法[12]等。其中高效液相色譜法多以甲醇-水、乙腈-水等為流動相體系,EPA甲酯和DHA甲酯的峰形對稱性較差,與相鄰色譜峰難以有效分離。因脂肪酸甲酯的紫外吸收較弱,選擇220 nm為檢測波長,基線噪音大,干擾峰較多,不適合多組分甲酯的測定[6]。質譜法成本較高,紅外光譜法難以準確定量,而三酰甘油沸點較高,氣相色譜法也很難直接測定。因此目前多以氣相色譜法測定脂肪酸甲酯表征對應三酰甘油的含量[13-20]。本文的研究中均以脂肪酸甲酯代替三酰甘油,建立可批量檢測、操作簡便、色譜分離完全、專屬性好的氣相色譜檢測方法,測定各脂肪酸甲酯,再計算三酰甘油的含量。
1 儀器與試藥
7820A氣相色譜儀(美國安捷倫公司),BT125D電子天平(德國賽多利斯公司),AUW220D電子天平(島津儀器有限公司)。
三氯甲烷、甲醇、無水氯化鈣均為分析純,采購自廣州化學試劑廠;正己烷采購自Honeywell,甲醇鈉(批號SHBC0411V)、2,6-二叔丁基甲基苯酚(以下簡稱BHT,批號P500117)、十七烷酸甲酯(批號BCBL1383V,純度99.7%)、辛酸甲酯(MKBN2148,純度99.9%)、癸酸甲酯(BCBS2886V,純度99.6%)、亞油酸甲酯(BCBG7530V,純度99.7%)、EPA甲酯(BCBL3641V,純度99.6%)、DHA甲酯(BCBL9396 V,純度99.6%),辛酸三酰甘油(BCBJ7208V,純度99.6%)、癸酸三酰甘油(SLBB6226V,純度100%)、亞油酸三酰甘油(SLBB9084V,純度98.0%)均采購自Sigma。魚油脂肪乳注射液(批號:15-1,15-2,15-3,廣東嘉博制藥有限公司),規格是250 mL∶50 g油脂∶3 g磷脂。原研藥(批號:120578083,德國貝朗)。
2 試驗方法
2.1 色譜條件
色譜柱:DB-WAX(30 m×0.25 mm,0.25 μm);FID檢測器溫度為275℃;進樣口溫度為260℃;載氣:高純氮氣(純度>99.99%),載氣流速:1 mL/min;分流比:10∶1;進樣量:1 μL。升溫程序:起始溫度為60℃,維持1 min,以每分鐘20℃的速率升至180℃,再以每分鐘4℃的速率升至240℃,保持18 min。
2.2 稀釋劑
取BHT約75 mg,精密稱定,溶于1 000 mL三氯甲烷和500 mL甲醇的混合溶液中,得到稀釋劑。
2.3 內標溶液的制備
取十七烷酸甲酯約100 mg,精密稱定,于50 mL容量瓶中,加入稀釋劑溶解并稀釋至刻度,得到內標儲備液(2 mg/L)。精密量取上述溶液10 mL,至100 mL量瓶中,加入稀釋劑稀釋至刻度,作為內標溶液。
2.4 供試品溶液的制備
精密量取魚油脂肪乳注射液100 μL,至20 mL容量瓶中,加入稀釋劑溶解并稀釋至刻度。精密量取上述溶液與內標液各1 mL,至具塞試管中,混勻,得到溶液A。在通氮氣的環境下,蒸干溶液A的溶劑,得到殘渣。
將上述殘渣溶于2 mL正己烷中,加入100 μL甲基化試劑(將1 mL甲醇鈉溶液于1.7 mL甲醇中),混勻并劇烈振蕩。放置30 min,加入約100 mg無水氯化鈣,搖勻并過濾,取續濾液作為供試品溶液。
2.5 混合對照品溶液的制備
分別取約120 mg辛酸三酰甘油,80 mg癸酸三酰甘油,90 mg亞油酸三酰甘油,16 mg EPA甲酯和11 mg DHA甲酯,精密稱定,至20 mL容量瓶中,加入稀釋劑溶解并定容,得到5種對照品儲備液。精密量取上述溶液各1 mL,置同一20 mL容量瓶中,加入內標儲備液2 mL,用稀釋劑稀釋至刻度,得到混合對照品溶液A。取1 mL此溶液,按照“2.4項下”,從“在通氮氣的環境下”開始同法操作,得到混合對照品溶液。
2.6 單個對照品溶液的制備
取“2.5”項下對照品儲備液各1 mL,分別置20 mL容量瓶中,加入稀釋劑稀釋至刻度,得到5種對照品溶液。取1 mL上述溶液與1 mL內標溶液,按照“2.4項下”供試品溶液,從“在通氮氣的環境下”開始同法操作,制備單個對照品溶液。
2.7 峰定位溶液的制備
另取辛酸甲酯、癸酸甲酯、亞油酸甲酯、EPA甲酯和DHA甲酯適量,加入稀釋劑溶解,作為峰定位溶液。
2.8 空白溶液的制備
取稀釋劑1 mL,加入1 mL內標溶液,按照“2.4”項下,從“在通氮氣的環境下”開始同法操作,制備空白溶液。
2.9 計算公式
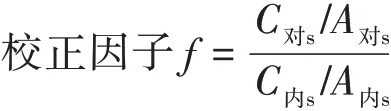
式中:A對s為對照品溶液中待測物的峰面積;C對s為對照品溶液待測物的濃度;A內s為對照品中內標的峰面積;C內s為內標的濃度。
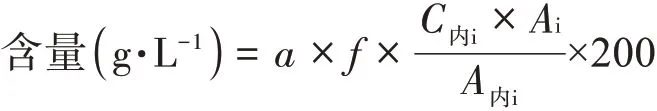
式中:f為校正因子;C內i為供試品溶液中內標的濃度;Ai為供試品溶液中待測組分的峰面積;A內i為供試品溶液中內標的峰面積;a為換算系數[M三酰甘油/(3×M甲酯)];M三酰甘油為三酰甘油型化合物的分子量(EPA三酰甘油=945.6/DHA三酰甘油=1023.6);M甲酯為甲酯型化合物的分子量(EPA甲酯=316.5/DHA甲酯=342.5)。
注:測定辛酸三酰甘油、癸酸三酰甘油和亞油酸三酰甘油的對照品均是三酰甘油對照品,計算時不需要換算系數,200為供試品的稀釋倍數。
3 方法學驗證內容
3.1 專屬性
取空白溶液、供試品溶液、混合對照品溶液、單個對照品溶液和峰定位溶液進樣,記錄色譜圖。
結果顯示:稀釋劑不干擾主成分峰的測定,供試品溶液中的各目標組分色譜峰能與單個對照品的色譜峰的保留時間一致。供試品溶液色譜圖中,目標組分色譜峰與相鄰色譜峰分離度不小于1.5。
3.2 線性和范圍
取“2.5”項下各對照品儲備液0.6、0.8、1.0、1.2、1.4 mL,置同一20 mL容量瓶中,加入內標儲備液2 mL,用稀釋劑稀釋至刻度。精密量取上述溶液1 mL,按“2.4”項下,從“在通氮氣的環境下”開始同法操作,得到系列濃度的混合對照品溶液,注入色譜儀。以濃度為橫坐標,各組分與內標峰面積比為縱坐標,得到線性回歸方程和相關系數。結果顯示,各對照品組分在各濃度范圍內線性關系良好。
3.3 進樣重復性
取“2.4”項下制備的供試品溶液,連續進樣6針,以峰面積計算RSD值,結果見表2。結果顯示,進樣6針各脂肪酸甲酯的峰面積RSD<1.0%,進樣重復性良好。
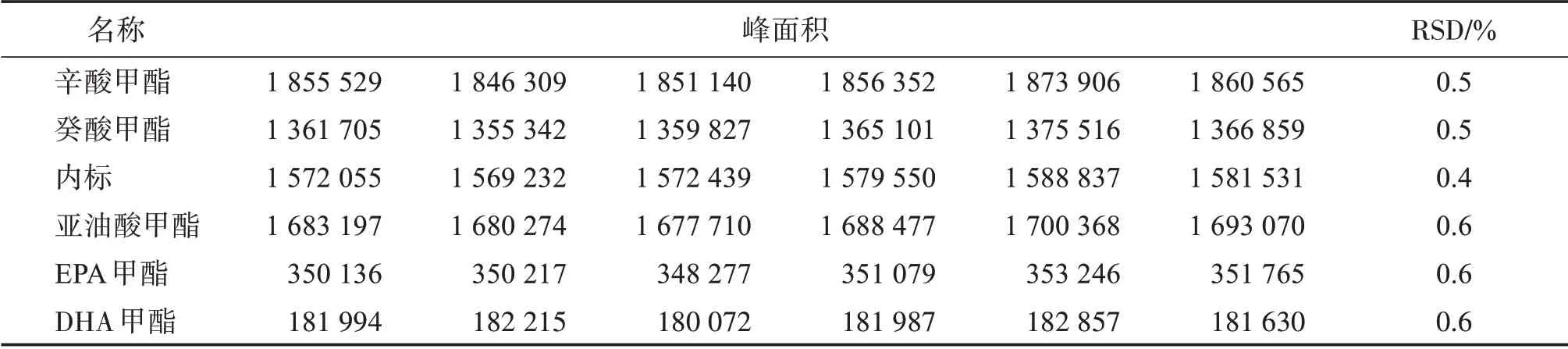
表2 進樣重復性Table 2 Repeatability of injections
3.4 重復性
按“2.4”項下方法,制備6份供試品溶液,注入色譜儀,記錄色譜圖,計算各甘油三酸酯含量和RSD,結果見表3。結果顯示,6份供試品中各三酰甘油的含量RSD<1.5%,供試品測定重復性良好。
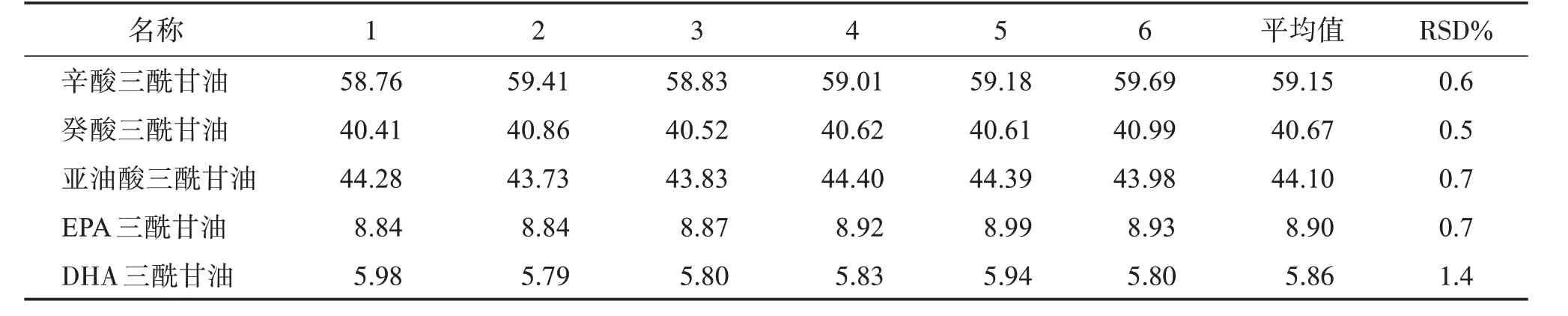
表3 重復性結果Table 3 Results of reproducibility ρ/(g·L-1)
3.5 中間精密度
由不同的實驗員在不同的日期,按“3.4”項下測定本品各三酰甘油含量,計算RSD。結果見表4。結果顯示,不同實驗人員測定各三酰甘油含量的RSD均小于2.0%,方法精密度良好。
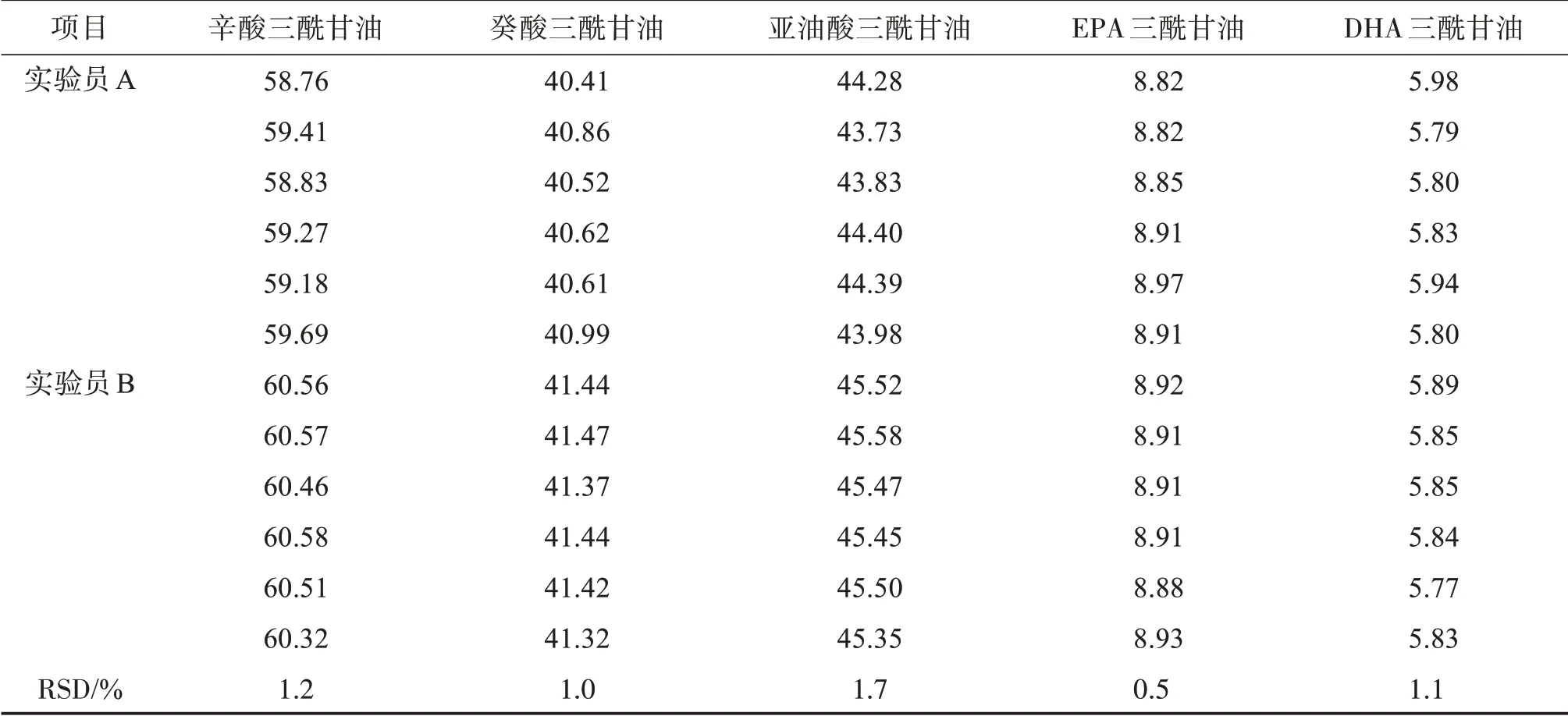
表4 中間精密度結果Table 4 Results of intermediate precision(n=12) ρ/(g·L-1)
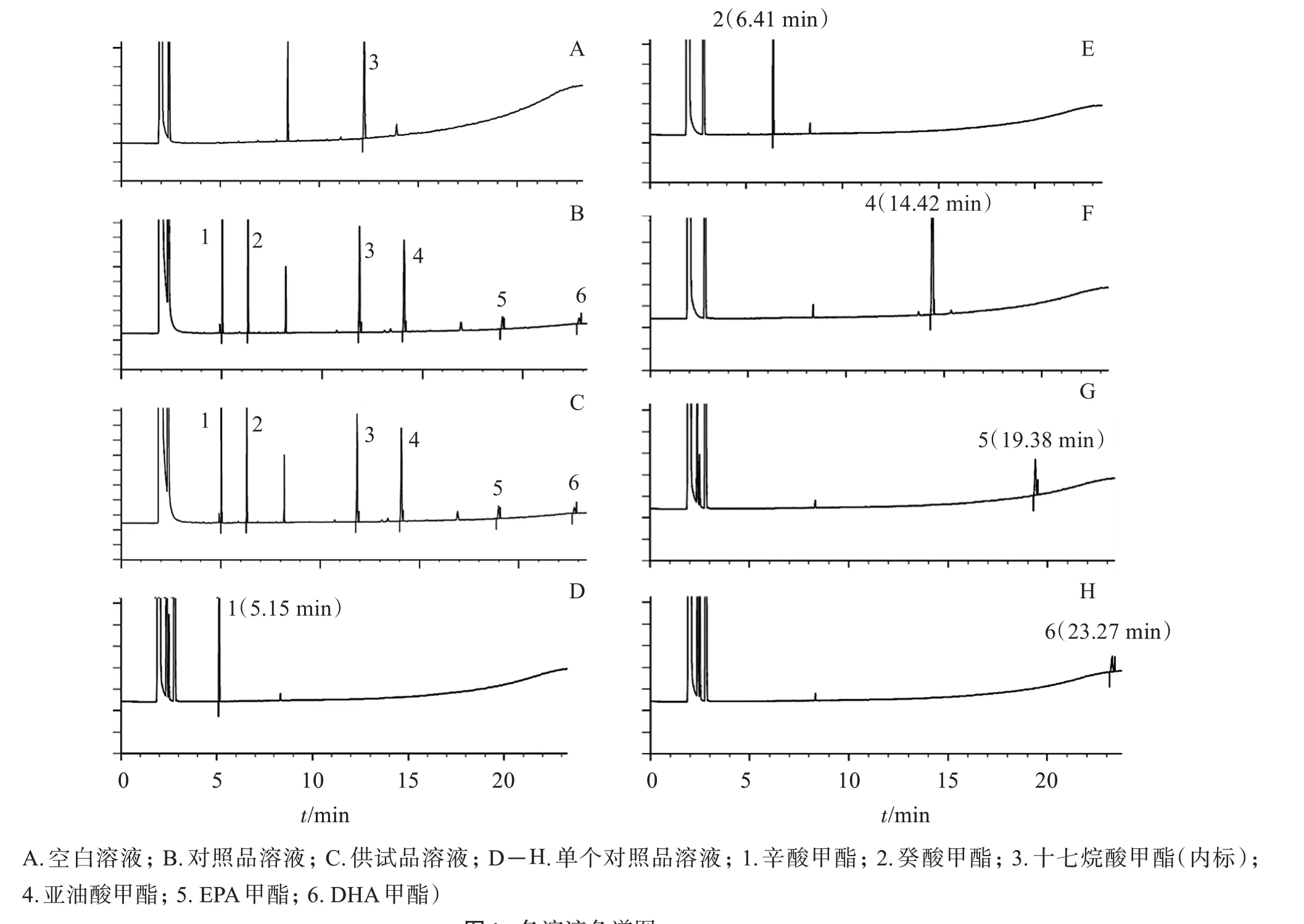
圖1 各溶液色譜圖Figure 1 Chromatograms of solutions
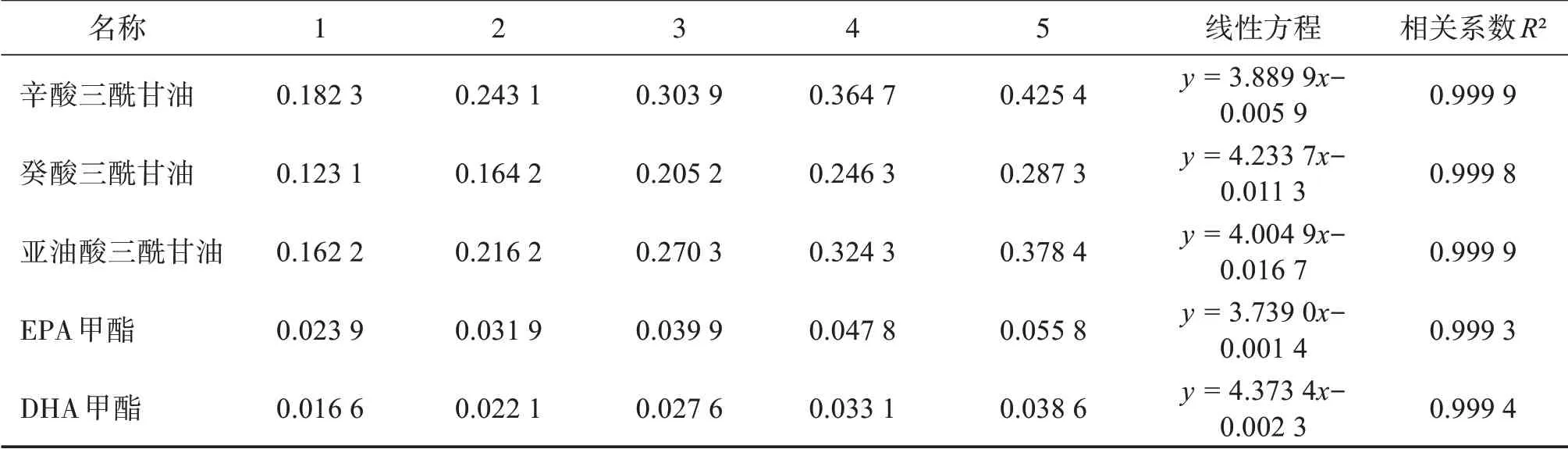
表1 線性試驗Table 1 Linear test ρ/(g·L-1)
3.6 回收率
取約150 mg辛酸三酰甘油,100 mg癸酸三酰甘油、112 mg亞油酸三酰甘油、20 mg EPA甲酯、14 mg DHA甲酯置同一50 mL量瓶中,精密稱定,加稀釋劑溶解并稀釋至刻度,得到對照品儲備液。
精密量取魚油脂肪乳注射液0.25 mL,共9份,分別置于100 mL容量瓶中,分別加入上述對照品儲備液3、5、7 mL(每個濃度制備3份溶液),用稀釋劑稀釋至刻度。得到相當于供試品濃度80%、100%、120%的溶液。分別精密取1 mL上述溶液和1 mL內標溶液,按照“2.4”項下同法處理。所得溶液注入色譜儀,記錄色譜圖,計算回收率和RSD。結果見表5,三酰甘油的平均回收率在99.4%~100.2%之間,RSD在1.3%~4.4%之間,方法的準確度良好。
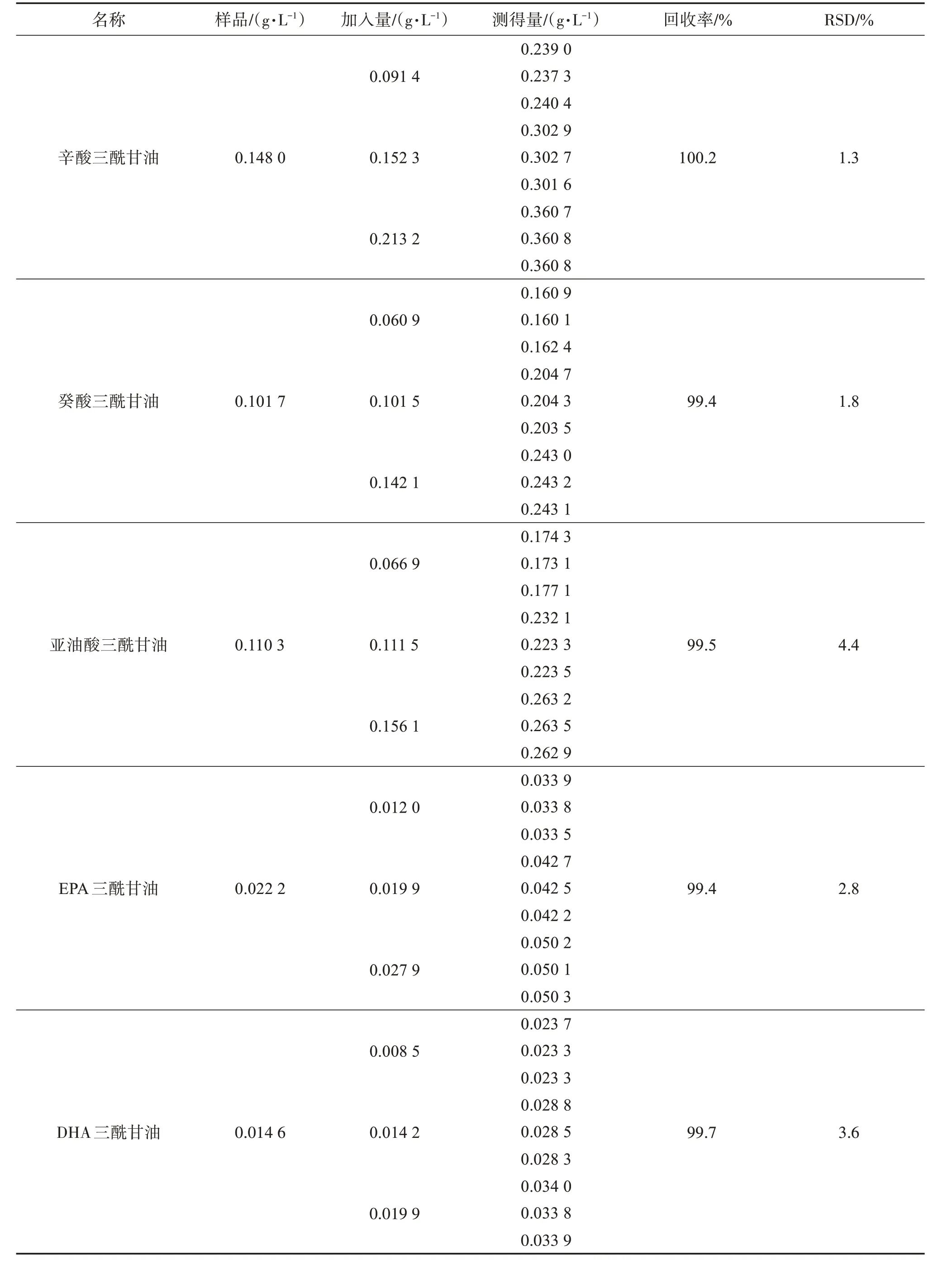
表5 回收率試驗結果Table 5 Results of recoveries
3.7 檢測限和定量限
取混合對照品溶液,通過逐級稀釋后進樣,使目標化合物峰信噪比值約為10∶1,作為定量限濃度。確定濃度后連續進樣6針,計算保留時間RSD。使目標化合物峰信噪比值約為3∶1,作為檢測限濃度。結果見表6。
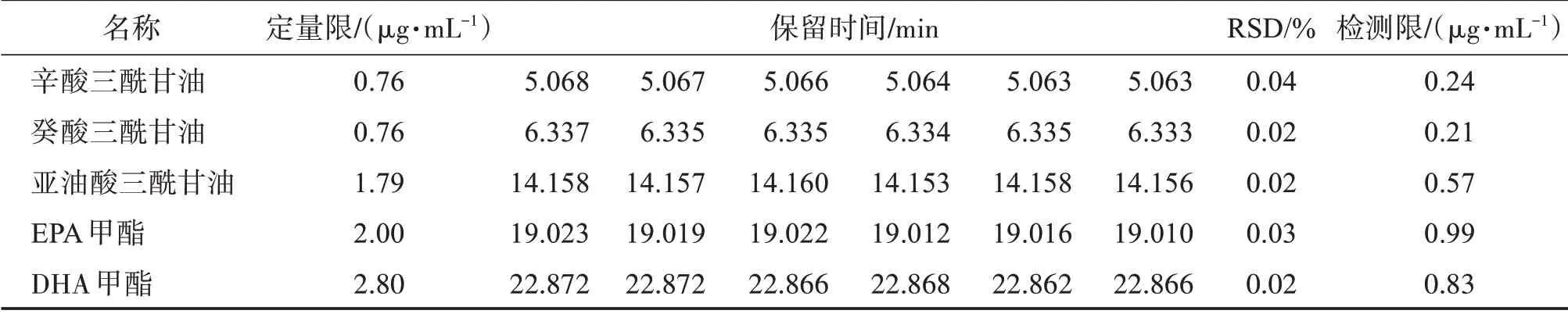
表6 定量限和檢測限結果Table 6 Results of LOQ and LOD
3.8 耐用性
3.8.1 供試品溶液穩定性取供試品溶液,分別于0、4、8、16、24、48 h進樣,以各脂肪酸甲酯與內標的峰面積比值計算RSD。結果見表7,供試品溶液放置48 h,峰面積比值RSD均小于1.0%,供試品溶液放置48 h穩定性良好。
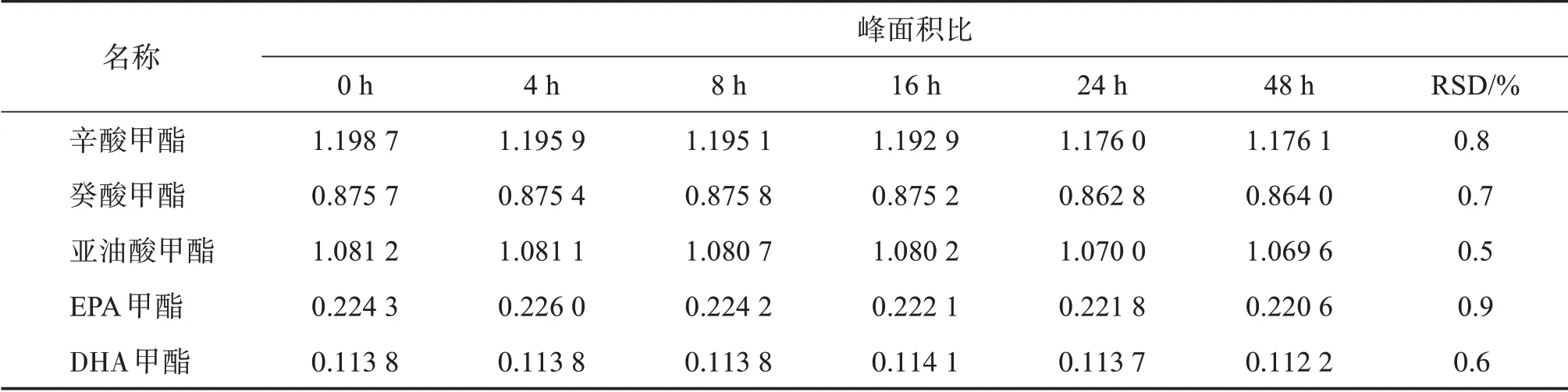
表7 供試品溶液穩定性結果Table 7 Results of solution stability
3.8.2 色譜條件變化取供試品溶液和混合對照品溶液,起始柱溫變化±5℃、載氣流速變化±20%,分別測定各三酰甘油的含量。結果見表8,起始柱溫變化±5℃、載氣流速變化±20%,對測定結果未見顯著影響。各三酰甘油的含量RSD均小于1.5%,方法耐用性良好。
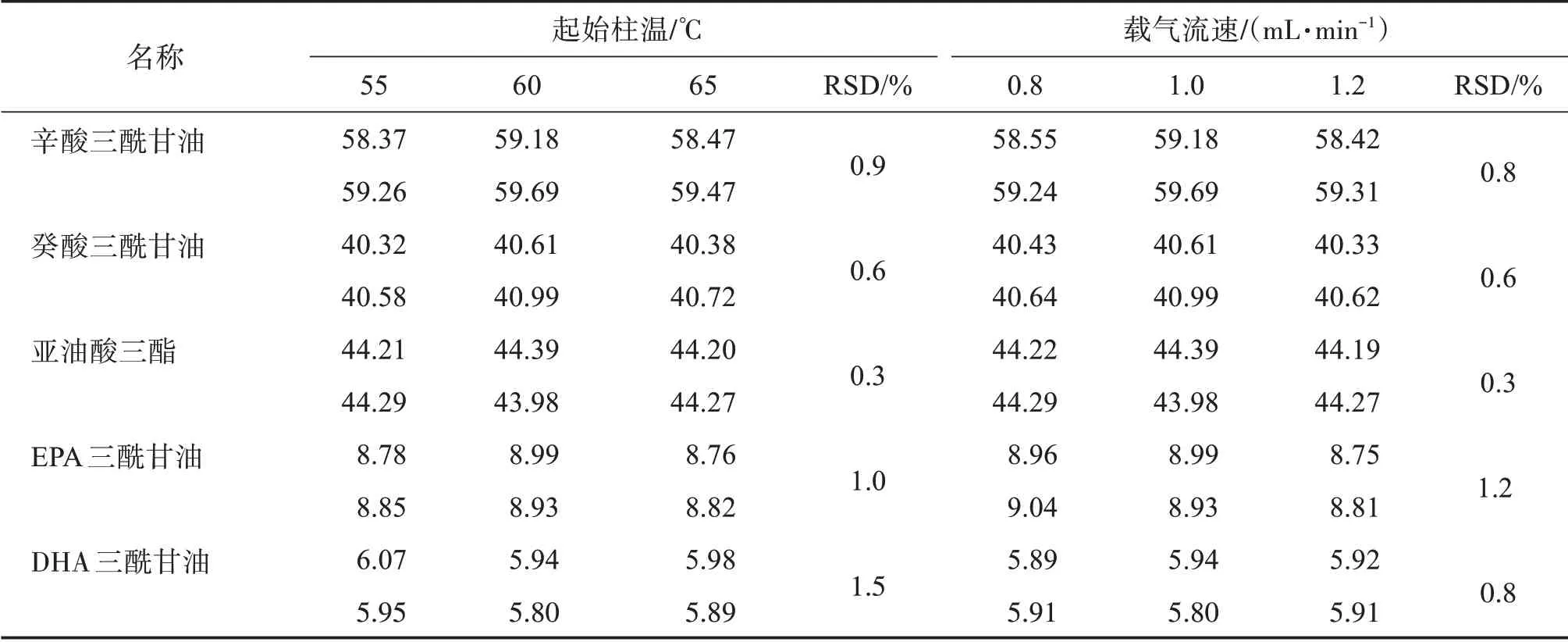
表8 耐用性結果Table 8 Results of robustnessρ(/g·L-1)
3.9 樣品測定
取本品3批和原研藥一批,按照“2試驗方法”項下,分別測定各三酰甘油的含量,結果見表9。結果顯示自研制劑的含量不低于原研藥,且符合本品擬定的內控標準規定:本品含中鏈三酰甘油(辛酸三酰甘油+癸酸三酰甘油)應為95.0~105.0 g/L,含大豆油(按亞油酸三酰甘油計)不得低于40 g/L,含EPA三酰甘油(C20∶5n-3)和DHA三酰甘油(C22∶6n-3)含量之和不得低于11.0 g/L。
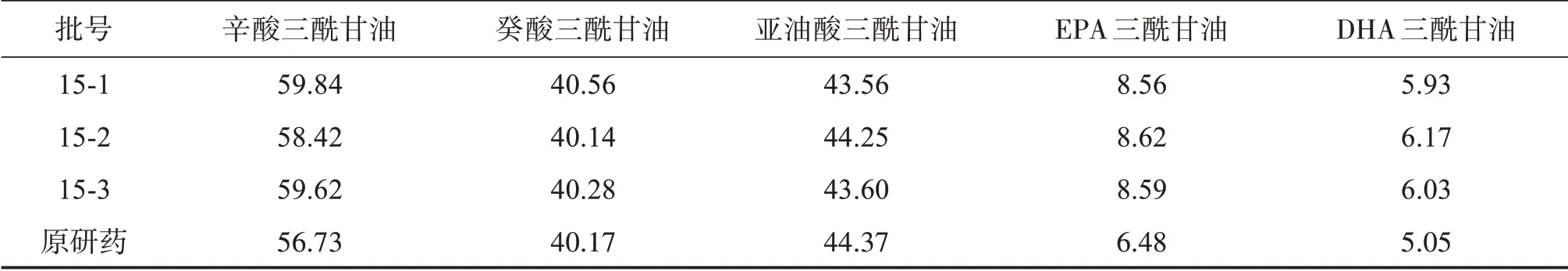
表9 樣品測定Table 9 Determination of samples ρ/(g·L-1)
4 討論
魚油富含ω-3多不飽和脂肪酸和三酰甘油,制備魚油注射液的油相多為大豆油、棕櫚油、卵磷脂等油脂類,油脂的主要成分為三酰甘油。氣相色譜法適合大多數的脂肪和脂肪酸,先把脂肪中的三酰甘油進行水解得到脂肪酸,脂肪酸經甲酯化得到脂肪酸甲酯,脂肪酸甲酯在FID檢測器和質譜檢測器上響應好,峰型對稱,塔板數高[13-14]。如果脂肪類樣品甲酯化不完全,樣品中可能會殘留三酰甘油、二酯和單酯,進入氣相色譜柱容易沉積在固定液表面,降低色譜柱的柱效。因此需要選擇方便、快捷的甲酯化方法。常用的甲酯化方法為酸催化和堿催化[15-18],包括三氟化硼甲醇法[21]、鹽酸甲醇法[22]、氫氧化鈉甲醇法[23]、乙酰氯甲醇法[24]。本文分別使用氫氧化鉀-甲醇,硫酸-甲醇加熱回流和甲醇鈉等方法進行試驗,對甲酯化效果進行比較,最終確定了以甲醇鈉作為甲酯化試劑,操作簡便,確保三酰甘油和脂肪酸全部轉化為甲酯。
本品經甲酯化后得到的亞油酸甲酯、EPA甲酯和DHA甲酯含雙鍵,EPA和DHA分別含有4個和5個活潑的亞甲基,此類亞甲基的存在使二者極易受到光、氧、高溫、自由基及金屬元素的影響,產生氧化、酸敗、聚合、雙鍵共軛等化學反應[19]。甲酯化過程需要經過水浴蒸干溶劑、甲基化反應、過濾等一系列操作,因此需要在樣品溶液中加入抗氧化劑保護不飽和脂肪酸。本研究參考歐洲藥典(EP)通則[20]選擇的BHT抗氧化能力強,在有機溶劑中的溶解性較好,在樣品處理中能有效防止不飽和脂肪酸的氧化。
本處方的輔料為磷脂、甘油、氫氧化鈉、水,以上輔料除卵磷脂可能含少量三酰甘油(不多于3%),蛋黃卵磷脂的處方量為1.2%,綜合評估均不干擾主成分的測定,因此本研究沒有做輔料干擾試驗。
本文采用直接進樣毛細管柱氣相色譜法測定魚油脂肪乳注射液中三酰甘油的含量,結果表明線性關系良好,方法重現性好,平均回收率在99.4%~100.2%之間,回收率高,可更好地進行產品的質量控制。
