QuEChERs法結合氣相色譜檢測大米中的有機磷農藥
2022-09-29 08:18:10關文碧朱國榕
肇慶學院學報 2022年5期
關鍵詞:殼聚糖
關文碧,朱國榕
(1.肇慶學院 食品與制藥工程學院,廣東 肇慶 526061;2.農業農村部農產品質量安全風險評估實驗室(肇慶),廣東 肇慶 526061)
水稻是我國第二大糧食作物,生長在溫暖、潮濕的環境中,雜草和病蟲害較多,農民為保證產量和品質常常會選擇施用農藥.若用藥不當,會影響大米的食用安全,危害人體健康,因此快速準確檢測大米中的農藥殘留對保障糧食安全和人體健康具有重要意義[1].有機磷農藥是常用的殺蟲殺螨劑,具有高效、低成本、控制范圍廣、植物毒性低等優點[2],但是大部分有機磷農藥對人和動物毒性較強,使用不當會導致哺乳動物急性中毒,損害機體.為防止產生食品安全危害,建立一種高效、準確、快速的檢測糧食中的有機磷農藥殘留量的方法非常重要.
有機磷農藥常用的檢測方法包括生物技術檢測法[3-5]、氣相色譜法[3-6]、氣質聯用法[7-8]、高效液相色譜法[9]、液相色譜串聯質譜法[10-11]等,其中氣相色譜搭配電子捕獲檢測器檢測有機磷農藥具有靈敏度高、專一性強、儀器價格較低、快速、高效等優點,被廣泛用于有機磷農藥的檢測[3-5,9].樣品前處理是農藥殘留分析的關鍵步驟之一,通過選擇合適的前處理方法能有效去除雜質,減少對檢測結果的干擾.目前,農藥殘留分析的樣品前處理技術主要包括固相萃取法[9]、加速溶劑萃取法[12]、凝膠滲透色譜法[13]和QuEChERs法[14]等,QuEChERs法是2003年由美國農業部首次提出,該方法具有簡便、快速,選擇性高等優點[14].本實驗采用改進的QuEChERs樣品前處理方法結合氣相色譜檢測大米中有機磷農藥殘留,并將該方法應用于肇慶地區市售大米中有機磷農藥殘留量檢測.
1 實驗部分
1.1 儀器與設備
GC-2014氣相色譜儀配備電子捕獲檢測器(日本島津儀器有限公司),CCA-20低溫冷卻循環水槽(鞏義市予華儀器有限責任公司),SHZ-D(Ⅲ)型循環水真空泵(上海邦西儀器科技有限公司),H1650R冷凍高速離心機(湖南湘儀實驗室儀器開發有限公司),TDL-40C低速大容量離心機(上海安亭科學儀器廠),BSA1245-CW電子天平(賽多利斯科學儀器北京有限公司),MX-S渦旋混勻儀(大龍興創實驗儀器北京股份公司).
色譜純乙腈(Macklin中國有限公司),色譜純乙酸乙酯(北京邁瑞達科技有限公司),蒸餾水(肇慶市鼎湖區飄雪飲用水有限公司),氯化鈉和無水硫酸鈉(分析純,廣州化學試劑廠),PSA(粒徑40~60 μm,上海安譜實驗科技股份有限公司),C18(粒徑40~75 μm,武漢瑞科美新能源有限公司),納米氧化鋁(純度99.99%、γ相、粒徑10 nm,阿拉丁試劑上海有限公司),低粘度殼聚糖、中粘度殼聚糖和高粘度殼聚糖(上海麥克林生化科技有限公司),弗羅里硅土(分粒度60~100目,山東西亞化學工業有限公司),乙酰甲胺磷標準品(純度99.9%,美國AccuStandard公司),樂果標準品(分析標準品,上海九鼎化學科技有限公司),馬拉硫磷標準品(純度99.15%,德國Dr.Ehrenstorfer GmbH公司),毒死蜱標準品(純度99.9%,美國AccuStandard公司).
1.2 標準溶液配制
999 mg∕L乙酰甲胺磷標準儲備液配制:稱取10 mg乙酰甲胺磷標準品于燒杯中,加入少量色譜純乙腈溶解,轉移到10 mL容量瓶并用色譜純乙腈定容至刻度線.1 230 mg∕L樂果標準儲備液配制:稱取12.3 mg樂果標準品于燒杯中,加入少量色譜純乙腈溶解,轉移到10 mL容量瓶并用色譜純乙腈定容至刻度線.1 200 mg∕L馬拉硫磷標準儲備液配制:稱取12.1 mg馬拉硫磷標準品于燒杯中,加入少量色譜純乙腈溶解,轉移到10 mL容量瓶并用色譜純乙腈定容至刻度線.999 mg∕L毒死蜱標準儲備液配制:稱取10 mg毒死蜱標準品于燒杯中,加入少量色譜純乙腈溶解,轉移到10 mL容量瓶并用色譜純乙腈定容至刻度線.10 mg∕L有機磷農藥混合標準溶液配制:分別吸取100 μL乙酰甲胺磷母液,81 μL樂果母液,83 μL馬拉硫磷母液,100 μL毒死蜱母液到10 mL容量瓶中,使用色譜純乙腈定容至刻度線.
1.3 樣品前處理
稱取5.00 g粉碎后的大米樣品置于50 mL塑料離心管中,加入5 mL蒸餾水和10 mL色譜純乙腈,渦旋提取2 min,加入2 g氯化鈉和2 g無水硫酸鈉,劇烈手搖30 s,于4 000 r∕min轉速下離心4 min.取8 mL上清液于雞心瓶中,旋蒸至干(水浴鍋不加熱),加入1.6 mL色譜純乙酸乙酯復容,取出1 mL復容液至2 mL裝有50 mg PSA和50 mg C18的離心管中,渦旋30 s,在10 565 r∕min轉速下離心3 min,取上清液過0.22 μm有機濾膜,待測.
1.4 儀器條件
色譜柱:SH-Rtx-5(30 m×0.25 mm,0.25 μm);載氣:高純氮氣,純度>99.99%;流速:1.5 mL∕min;進樣口溫度:270℃;檢測器溫度:300℃;進樣方式:分流進樣,分流比為15:1;進樣量:5 μL;程序升溫:初始溫度為120℃,以25℃∕min的速率升溫至170℃,繼續以10℃∕min的速率升溫至260℃,最后以5℃∕min的速率升溫至280℃,保留2 min,共運行17 min.乙酰甲胺磷的保留時間是4.1 min,樂果的保留時間是6.6 min,馬拉硫磷的保留時間是8.7 min,毒死蜱的保留時間是9.0 min.
2 結果與討論
2.1 凈化劑種類的優化
為了探究凈化劑的種類對農藥添加回收率R的影響,保持其他實驗條件不變,固定50 mg C18的前提下,搭配50 mg的PSA、納米氧化鋁、弗羅里硅土、低粘度殼聚糖、中粘度殼聚糖和高粘度殼聚糖.結果表明選用納米氧化鋁和弗羅里硅土時,乙酰甲胺磷的回收率低于70%,不滿足農藥殘留準則中規定的添加回收率范圍.選用PSA、低粘度殼聚糖、中粘度殼聚糖和高粘度殼聚糖時4種有機磷農藥的回收率分別為76.22%~127.58%、84.46%~104.51%、79.26%~100.30%和77.99%~103.28%,農藥殘留分析均滿足要求(圖1).圖2是PSA凈化后和低粘度殼聚糖凈化后的大米樣品提取凈化液色譜圖,橫坐標為時間,縱坐標為色譜峰響應值.從圖2的方框區域可看出PSA凈化的色譜圖中雜峰數量比低粘度殼聚糖凈化的色譜圖少,且PSA凈化后的提取溶液中雜峰的峰面積更小.由此可知,PSA的凈化效果更佳.綜合考慮,選擇PSA搭配C18為凈化劑.
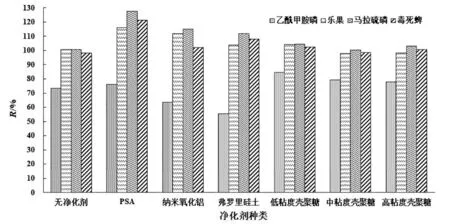
圖1 凈化劑種類對有機磷農藥回收率的影響
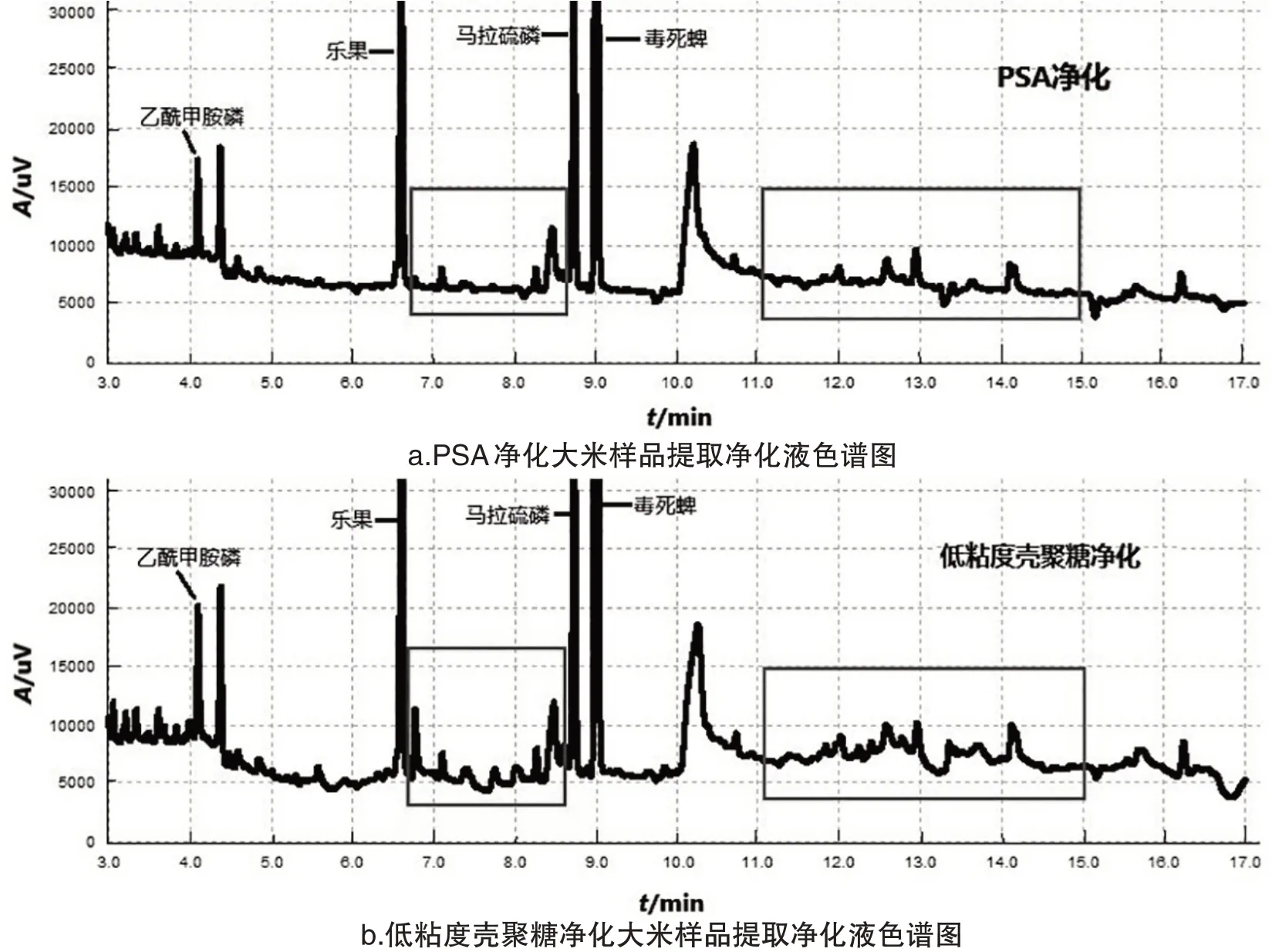
圖2 大米樣品提取凈化液色譜對比圖
2.2 凈化劑用量的優化
為了達到更好的凈化效果,本實驗固定C18的用量為50 mg,設置6個PSA用量(50 mg、60 mg、70 mg、80 mg、90 mg、100 mg)進行優化,實驗結果見圖3.當PSA使用量為60~100 mg時,乙酰甲胺磷的回收率低于70%,不滿足農藥殘留分析要求;PSA使用量為50 mg時,4種有機磷農藥的回收率在76.22%~116.30%之間,因此選擇50 mg PSA和50 mg C18作為此次實驗最優的凈化劑用量.
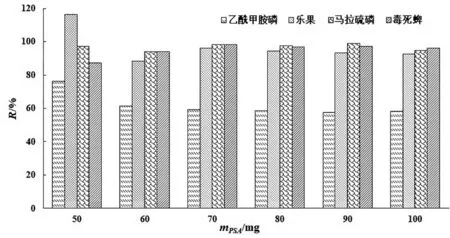
圖3 PSA用量對有機磷農藥回收率的影響
2.3 方法的確證
2.3.1 線性范圍
分別用色譜純乙酸乙酯和空白大米樣品提取凈化液稀釋10 mg∕L有機磷農藥混合標準溶液,得到0.01 mg∕L、0.02 mg∕L、0.05 mg∕L、0.1 mg∕L、0.2 mg∕L、0.5 mg∕L、1 mg∕L、2 mg∕L的溶劑標準系列溶液和基質標準系列溶液,進GC-ECD分析.以有機磷農藥濃度作為橫坐標,有機磷農藥峰面積為縱坐標,繪制標準曲線,并分別求出溶劑標準曲線和基質標準曲線,結果如表1所示.4種有機磷農藥在0.01~2 mg∕L的范圍內線性關系良好,相關系數均大于0.992 3,能滿足定量分析的需求.
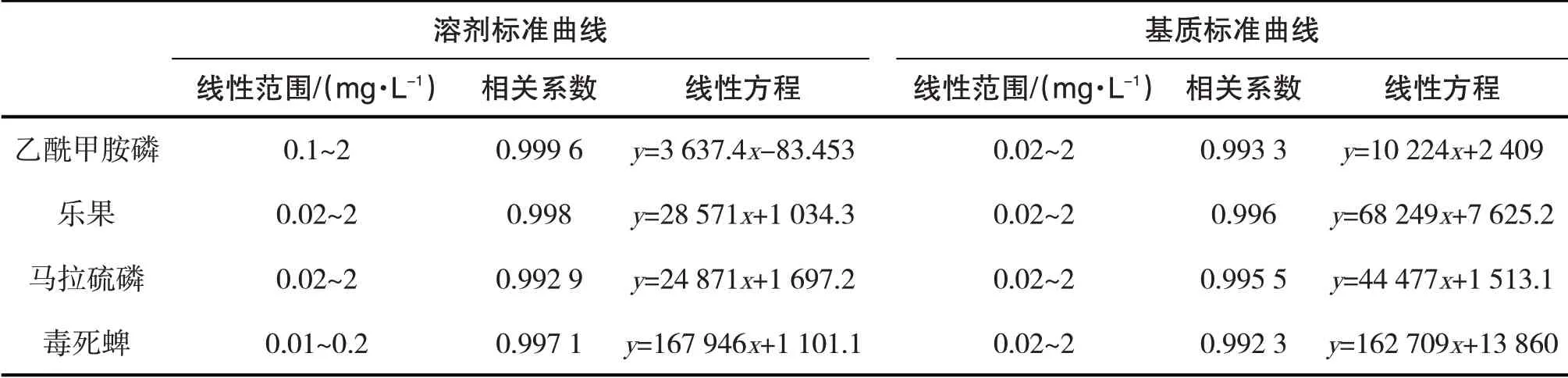
表1 有機磷農藥的溶劑標準曲線和基質標準曲線
2.3.2 基質效應
基質效應(Matrix effect,ME)是指樣品分析液中除目標分析物之外的成分使分析物的響應值發生變化,具體表現為以純溶劑配制的農藥標準品和以樣品基質配制的農藥標樣中的相同組分的檢測值存在差異,從而影響了分析的準確度.通過如下公式判斷本實驗中其他組分產生的基質效應對待測物質的影響程度:
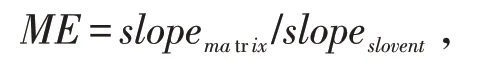
式中:slopematrix為基質標準曲線斜率,slopeslovent為溶劑標準曲線斜率.
通過計算,乙酰甲胺磷的ME為2.72,樂果的ME為2.37,馬拉硫磷的ME為1.81,毒死蜱的ME為1.60.因為ME>1.1,所以4種農藥均表現為明顯增強的基質效應.
2.3.3 準確度和精密度
按照上述所說的前處理方法和檢測方法,以及實驗得到的最佳條件,在4個不同的添加濃度(0.01 mg∕kg、0.05 mg∕kg、0.1 mg∕kg、0.5 mg∕kg)下進行添加回收實驗,每個濃度各做5個平行實驗,結果如表2所示,4種有機磷農藥的平均回收率在70.95%~113.49%之間,相對標準偏差RSDs在1.05%~19.10%之間,滿足農藥殘留分析的準確度要求.
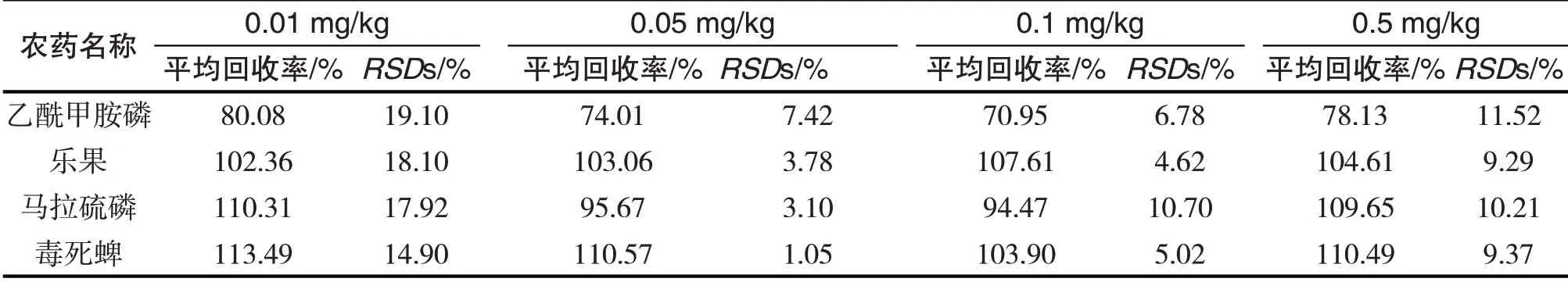
表2 4種有機磷農藥的平均回收率和相對標準偏差
2.3.4 檢出限和定量限
方法的靈敏度由方法的檢出限和定量限進行評價.本實驗在空白大米提取凈化液中添加有機磷農藥的標準溶液,進GC-ECD檢測,選取使檢測系統產生3倍噪音信號所需待測物質的濃度作為檢出限[15],4種有機磷農藥的檢出限均為0.02 mg∕L.基于方法準確度和精密度的實驗結果,在滿足回收率為70%~120%和RSDs≤20%的前提下,本實驗選取添加回收實驗中最低添加濃度作為方法的定量限[15],本方法中4種有機磷農藥的定量限均為0.01 mg∕kg.
2.3.5 實際樣品的測定
基于本方法對肇慶市端州區銷售的15種大米中的乙酰甲胺磷、樂果、馬拉硫磷、毒死蜱的殘留量進行檢測分析,檢測結果如表3所示.15種市售大米中,樂果和毒死蜱的檢出率均為0%,殘留量低于方法的定量限;乙酰甲胺磷的檢出率為6.67%,殘留量為0.21 mg∕kg;馬拉硫磷的檢出率為100%,殘留量為0.02~0.09 mg∕kg.本實驗結果表明肇慶地區市售食用大米中乙酰甲胺磷、樂果、馬拉硫磷、毒死蜱的殘留量均低于GB2763-2019食品安全國家標準《食品中農藥最大殘留限量》(表4).

表3 15種市售大米的4種有機磷農藥殘留量
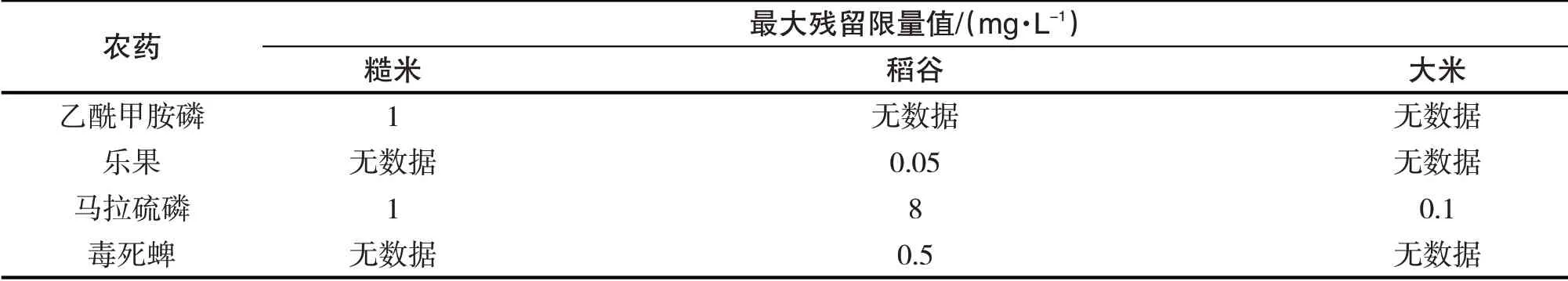
表4 國標中4種有機磷農藥的最大殘留限量值
3 結論
本試驗采用改進的QuEChERS樣品前處理方法,結合氣相色譜技術建立了大米中乙酰甲胺磷、樂果、馬拉硫磷、毒死蜱4種有機磷農藥的QuEChERS-GC-ECD殘留檢測方法.實驗中優化了凈化劑的種類和用量,線性范圍在0.01~2 mg∕L濃度范圍內,乙酰甲胺磷、樂果、馬拉硫磷、毒死蜱顯現良好的線性關系,相關系數均大于0.992 3.在0.01、0.05、0.1、0.5 mg∕kg4個添加濃度下進行大米中有機磷農藥的添加回收實驗,4種有機磷農藥的平均添加回收率為70.95%~113.49%,RSDs為1.05%~19.10%,方法的檢出限為0.02 mg∕L,定量限為0.01 mg∕kg.將該方法應用于市售15種大米中有機磷農藥的殘留量檢測,結果表明,實際樣品中有機磷農藥殘留量均低于國家標準.該方法的前處理步驟簡單,操作性強,準確度和精密度較高,可用于大米中4種有機磷農藥的殘留檢測.
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
