阿卡波糖口服固體制劑含量測定方法的優(yōu)化
2022-09-13 08:24:02石笑弋易必新李昌亮李帥劉雁鳴蘭文李健和湖南省藥品檢驗(yàn)檢測研究院長沙4000國家藥品監(jiān)督管理局藥用輔料工程技術(shù)研究重點(diǎn)實(shí)驗(yàn)室長沙4000中南大學(xué)湘雅二醫(yī)院藥學(xué)部長沙400
中南藥學(xué) 2022年6期
關(guān)鍵詞:方法
石笑弋,易必新*,李昌亮,2,李帥,2,劉雁鳴,2,蘭文,2,李健和(.湖南省藥品檢驗(yàn)檢測研究院,長沙 4000;2.國家藥品監(jiān)督管理局藥用輔料工程技術(shù)研究重點(diǎn)實(shí)驗(yàn)室,長沙 4000;.中南大學(xué)湘雅二醫(yī)院藥學(xué)部,長沙 400)
阿卡波糖是德國拜耳公司于20 世紀(jì)80年代從一種游離放線菌發(fā)酵分離得到的產(chǎn)物,由4 個(gè)單糖構(gòu)成的寡糖,是世界上第一個(gè)α
-葡萄糖糖苷酶抑制劑類的口服降糖藥,主要用于2 型糖尿病的治療,可通過抑制淀粉酶來減少葡萄糖的吸收,更適合于降低以碳水化合物為主食的亞洲糖尿病患者的血糖。阿卡波糖口服固體制劑自1994年在中國批準(zhǔn)上市以來,不良反應(yīng)相對較小,市場占有率較高,應(yīng)用前景較廣。目前,國內(nèi)現(xiàn)有阿卡波糖及其制劑質(zhì)量標(biāo)準(zhǔn)收載于《中國藥典》2020年版二部,其他相關(guān)質(zhì)量標(biāo)準(zhǔn)及文獻(xiàn)中阿卡波糖含量測定的分析方法多采用HPLC 法,檢測器普遍應(yīng)用紫外或示差折光檢測器,亦有利用電噴霧檢測器等新型檢測器檢測,色譜柱大多使用氨基柱,流動(dòng)相均采用大量有機(jī)試劑。現(xiàn)有方法大多存在受環(huán)境影響基線波動(dòng)較大、靈敏度降低、柱效下降快或普及率不高等問題。因此,對阿卡波糖片及膠囊的含量測定方法進(jìn)行改進(jìn)十分必要。本文采用SCX 色譜柱,優(yōu)化了阿卡波糖口服固體制劑中阿卡波糖含量測定的方法,現(xiàn)報(bào)道如下。
1 儀器與試藥
賽默飛Dionex UltiMate 3000 高效液相色譜儀、PDA 紫外檢測器;十萬分之一XP205 電子分析天平(Mettler Toledo)。阿卡波糖對照品(批號:100808-201905,含量:99.4%,中國食品藥品檢定研究院,水分:3.45%)。阿卡波糖原料藥(杭州中美華東制藥有限公司,批號:1909156AK)。1個(gè)廠家的阿卡波糖膠囊及6 個(gè)廠家的阿卡波糖片共165 批次樣品。磷酸二氫鉀、氫氧化鈉、鹽酸等試劑為分析純(國藥集團(tuán)化學(xué)試劑有限公司);純水(Thermo Scientific GenPure 超純水機(jī)制備)。
2 方法與結(jié)果
2.1 色譜條件
色譜柱Welch Ultimate XB-SCX 柱(4.6 mm×250 mm,3 μm),流動(dòng)相0.02 mol·L磷酸二氫鉀溶液,流速1.0 mL·min,柱溫40℃,檢測波長210 nm,進(jìn)樣量10 μL。
2.2 溶液的配制
2.2.1 供試品溶液的配制 取本品20 片(或20粒內(nèi)容物),精密稱定,研細(xì),精密稱取適量(約相當(dāng)于阿卡波糖50 mg),置50 mL 量瓶中,加水適量,超聲使溶解并稀釋至刻度,搖勻,濾過,取續(xù)濾液作為供試品溶液。
2.2.2 對照品溶液的配制 取阿卡波糖對照品適量,加水制成每1 mL 中約含1 mg 的溶液作為對照品溶液。
2.2.3 陰性樣品溶液的配制 按處方比例及制備工藝,制備缺阿卡波糖的樣品,按“2.2.1”項(xiàng)下方法制備陰性樣品溶液。
2.2.4 系統(tǒng)適用性溶液的配制 取阿卡波糖原料藥約200 mg,置10 mL 量瓶中,加少量水使溶解,加0.1 mol·L氫氧化鈉溶液1 mL,混勻,室溫放置1 h,加0.1 mol·L鹽酸溶液1 mL,加水稀釋至刻度,搖勻,作為系統(tǒng)適用性溶液。
2.3 系統(tǒng)適用性試驗(yàn)
精密量取“2.2”項(xiàng)下陰性樣品溶液、系統(tǒng)適用性溶液及供試品溶液各10 μL,進(jìn)樣測定,結(jié)果主峰與相鄰雜質(zhì)峰均有效分離,以阿卡波糖色譜峰計(jì),拖尾因子小于1.2,理論板數(shù)大于5000。溶劑、輔料對測定均無干擾,見圖1。
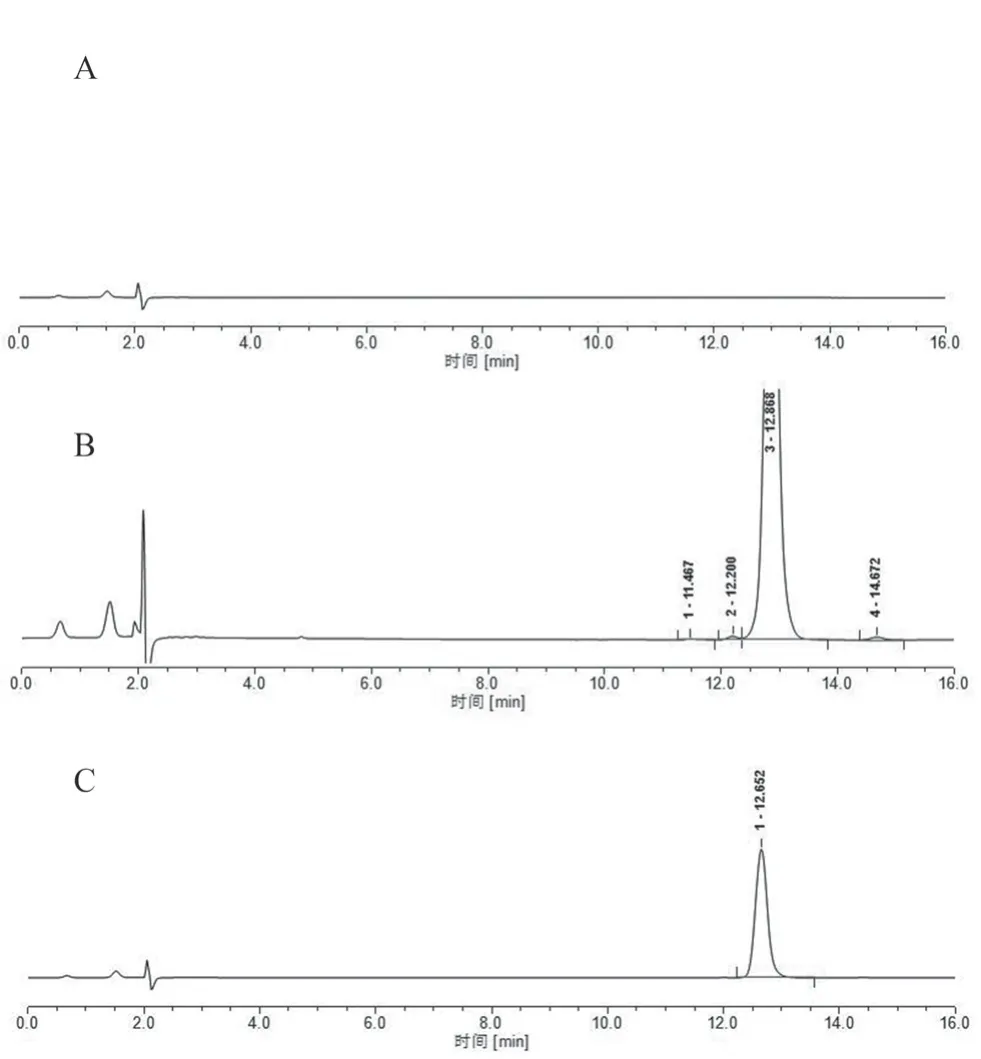
圖1 系統(tǒng)適用性試驗(yàn)HPLC 圖Fig 1 HPLC chromatogram of system suitability test
2.4 線性范圍、檢測限與定量限
精密稱取阿卡波糖對照品215.19 mg,置50 mL 量瓶中,加水適量,超聲使溶解并稀釋至刻度,搖勻,作為線性溶液①,取該線性溶液6、3、2.5、2、0.5、0.2 mL,分別置10 mL 量瓶中,加水稀釋至刻度,搖勻,得到質(zhì)量濃度為2.478、1.239、1.032、0.8260、0.2065、0.082 60 mg·mL的線性溶液②~⑦。進(jìn)樣測定,記錄色譜圖。以質(zhì)量濃度(X
)為橫坐標(biāo)和峰面積(Y
)為縱坐標(biāo)進(jìn)行線性回歸,得阿卡波糖回歸方程為Y
=2.529×10X
-244.2,相關(guān)系數(shù)為1.000,線性范圍為0.082 60 ~4.130 mg·mL;并根據(jù)信噪比S/N
≈ 3 確定檢測限(LOD),S/N
≈ 10 確定定量限(LOQ),結(jié)果檢測限為6.8 μg·mL,定量限為22 μg·mL。2.5 精密度試驗(yàn)
取“2.4”項(xiàng)下質(zhì)量濃度約為1.0 mg·mL的對照品溶液,連續(xù)進(jìn)樣6 次,記錄保留時(shí)間與峰面積。結(jié)果阿卡波糖峰面積的RSD
<1.5%,保留時(shí)間的RSD
均<1.0%,表明儀器精密度良好。2.6 重復(fù)性試驗(yàn)
取同一批樣品(阿卡波糖片,批號:210301,規(guī)格:50 mg)6 份,按“2.2.1”項(xiàng)下方法制備并測定,結(jié)果含量平均值為100.11%,RSD
為0.70%,表明方法重復(fù)性良好。2.7 溶液穩(wěn)定性試驗(yàn)
取“2.5”項(xiàng)下溶液室溫避光放置,分別于0、2、4、8、12、16、20 及24 h 進(jìn)樣考察,阿卡波糖的峰面積RSD
均<2.0%,表明溶液在24 h 內(nèi)穩(wěn)定性良好。2.8 回收試驗(yàn)
取陰性樣品,精密加入阿卡波糖對照品,制成濃度相當(dāng)于阿卡波糖含量100%水平的供試溶液共6 份,進(jìn)樣分析,記錄色譜圖,按外標(biāo)法以峰面積計(jì)算阿卡波糖的含量,結(jié)果平均回收率為99.41%,RSD
為0.32%(n
=6)。2.9 耐用性試驗(yàn)
通過調(diào)節(jié)流速及柱溫,考察阿卡波糖峰的變化情況。結(jié)果阿卡波糖峰的理論板數(shù)均大于5000,拖尾因子均小于1.2,與相鄰雜質(zhì)峰均能有效分離。
2.10 樣品測定
取165 批次樣品,按“2.2.1”項(xiàng)下方法制備供試品溶液,按“2.1”項(xiàng)下色譜條件進(jìn)行測定,按外標(biāo)法以峰面積計(jì)算,并將含量測定結(jié)果與《中國藥典》2020年版二部收載的采用氨基柱測定的結(jié)果進(jìn)行比較,采用SPSS 統(tǒng)計(jì)軟件對兩種方法含量測定測結(jié)果進(jìn)行配對樣品t
檢驗(yàn)。結(jié)果相關(guān)系數(shù)r
=0.542,P
<0.001,認(rèn)為兩配對變量有相關(guān)關(guān)系;t
=-3.263,d
=164,雙尾檢驗(yàn)概率P
=0.001,表明兩種檢驗(yàn)方法所測結(jié)果具有顯著性差異,結(jié)果見表1。表1 樣品中阿卡波糖的含量結(jié)果( =165)
Tab 1 Content of acarbose in the sample ( =165)
含量均值的標(biāo)準(zhǔn)差/%藥典(采用NH2 色譜柱)98.811.12640.0877本文(采用SCX 色譜柱)99.091.21600.0946測定方法含量均值/%標(biāo)準(zhǔn)差/%
3 討論
《中國藥典》2020年版二部是采用HPLC 法測定阿卡波糖的含量。采用氨基色譜柱,以乙腈-磷酸鹽緩沖液(75∶25)為流動(dòng)相,流速2 mL·min,柱溫35℃,檢測波長 210 nm。該方法存在很多缺點(diǎn),嚴(yán)重影響了方法的重現(xiàn),并造成應(yīng)用困難:① 相鄰降解雜質(zhì)Ⅰ峰與主峰的分離效果不佳,只有系統(tǒng)適用性中規(guī)定雜質(zhì)Ⅰ的峰高(Hp)與雜質(zhì)Ⅰ和阿卡波糖兩峰之間的峰谷(Hv)之比Hp/Hv不得低于2.0 的要求;② 氨基柱耐用性較差,鍵合相容易流失,最多使用150 次左右,柱效嚴(yán)重降低,色譜峰峰形變差,影響準(zhǔn)確定量;③ 流動(dòng)相中有機(jī)試劑毒性大,流速大,檢測成本高。因此,有必要優(yōu)化阿卡波糖含量測定的分析方法,改善方法的重現(xiàn)性和可應(yīng)用性,提高藥品質(zhì)量控制水平。目前尚未見采用SCX 色譜柱HPLC 法測定阿卡波糖含量的報(bào)道。
3.1 流動(dòng)相的選擇
阿卡波糖在水中極易溶解,在乙腈中不溶。本文采用水為溶劑,0.02 mol·L磷酸二氫鉀溶液為流動(dòng)相,能解決《中國藥典》2020年版二部方法中因溶劑與流動(dòng)相中有機(jī)相比例差異對靈敏度、出峰時(shí)間及峰形等的影響。
3.2 色譜條件的優(yōu)化
本文以0.02 mol·L磷酸二氫鉀溶液為流動(dòng)相,流速1.0 mL·min,柱溫40℃的色譜條件,采用SCX 色譜柱(4.6 mm×250 mm,3 μm),系統(tǒng)適用性要求均能達(dá)到。SCX 色譜柱是一種典型的陽離子交換柱,以磺酸基強(qiáng)陽離子硅膠為鍵合相,用于堿性、水溶的化合物的分離。SCX 色譜柱較氨基色譜柱的固定相穩(wěn)定,不易產(chǎn)生柱流失,在使用約300 次后,組分仍然能保持良好的峰形,對稱性好,拖尾因子小,分離度與理論板數(shù)能滿足系統(tǒng)適用性要求。
3.3 小結(jié)
本文采用SCX 色譜柱的HPLC 法測得的阿卡波糖口服固體制劑含量結(jié)果準(zhǔn)確、方法可靠,與《中國藥典》2020年版收載的方法相比,該方法毒性小、成本低、專屬性強(qiáng)、色譜柱耐用性好,具備一定的實(shí)用性。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(bào)(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
