鋁摻雜對高鎳無鈷LiNi0.95Mn0.05O2正極材料結構與性能的影響①
2022-09-09 05:18:24李靈均曾愛香周友元
礦冶工程 2022年4期
王 楚,李靈均,曾愛香,譚 磊,周友元
(1.長沙理工大學 材料科學與工程學院,湖南 長沙 410000;2.長沙理工大學 能源與動力工程學院,湖南 長沙 410000;3.湖南長遠鋰科股份有限公司,湖南 長沙 410000)
提高層狀氧化物正極材料中的Ni含量可以進一步提升電池的能量密度[1?2]。此外,由于Co元素價格昂貴以及導致環境污染等問題,發展層狀高鎳無鈷正極材料已成為一種趨勢[3]。LiNi0.95Mn0.05O2正是這樣一類具有高比容量、低成本和低污染的高鎳無鈷層狀正極材料。然而,Co元素的去除也會引起一些問題,主要包括:①材料的Li+/Ni2+混排程度增加[4];②材料在充放電過程中的相變程度加劇,在材料中造成應力集中并引發微裂紋的產生[5?6]。通過元素摻雜來進一步提升LiNi0.95Mn0.05O2材料的結構穩定性以及電化學性能是非常有效的方法。本文采用固相燒結法合成了Al摻雜的LiNi0.95Mn0.05O2正極材料,并通過結構分析方法和電化學測試手段研究Al3+對該正極材料晶體結構和電化學性能的影響。
1 實驗部分
1.1 實驗原料及方法
LiNi0.95Mn0.05O2(簡稱NM)的制備:將Ni0.95Mn0.05(OH)2前驅體和LiOH·H2O按物質的量比Li∶[Ni+Mn]=1.05∶1稱量,然后置于研缽中研磨30 min,使其均勻混合,最后轉移至管式爐中480℃預燒5 h,再以5℃/min的升溫速率升至720℃煅燒10 h,待自然冷卻至室溫后即得到目標產物。
1%鋁摻雜LiNi0.95Mn0.05O2樣品(簡稱NM?A)的制備:將Ni0.95Mn0.05(OH)2前驅體、LiOH·H2O以及Al(OH)3按物質的量比Li∶[Ni+Mn]∶Al=1.05∶0.99∶0.01稱量,再采用與NM樣品相同的研磨方法和燒結制度獲得目標產物。
1.2 材料表征
采用X射線衍射儀(XRD,D8 Advance,Bruker)分析樣品晶體結構,測試范圍10°~90°,掃描速度5°/min。采用場發射掃描電子顯微鏡(SEM,LSM 7900F,JEOL)觀察樣品微觀形貌及元素分布。
1.3 電化學性能測試
正極極片中活性物質、乙炔黑和聚偏二氟乙烯(PVDF)的質量比為8∶1∶1,極片直徑12 mm,負載量2.2 mg/cm2。在充滿氬氣的手套箱中,以鋰金屬為負極組裝成CR2025型紐扣電池,電解液由1 mol/L LiPF6溶解在DMC、EMC和EC按體積比1∶1∶1混合的溶劑中構成。電化學性能測試在Neware電池測試系統完成,測試電壓范圍2.7~4.3 V(vs Li/Li+),溫度30℃。電化學阻抗譜(EIS)通過電化學工作站(CHI 660E)測試,頻率范圍10-3~105Hz,電壓振幅5 mV。
2 實驗結果與討論
2.1 晶體結構表征
圖1為原始樣品NM與摻雜樣品NM?A的XRD圖譜。2個樣品的XRD衍射峰都與標準PDF卡片一一對應,峰型都比較尖銳,均為六方晶系α?NaFeO2層狀結構,屬于R?3m空間群,且未觀察到明顯的雜質相,因此可以認為Al成功地摻入到LiNi0.95Mn0.05O2正極材料晶格中,而不是以第二相形式存在。此外,(006)/(102)峰都分裂比較明顯,表明所合成樣品具有良好的層狀結構,且結晶度較高[7]。不同的是,NM?A正極材料的(003)/(104)峰強比為1.95,高于NM樣品的峰強比1.93。由于Ni2+與Li+具有相似的離子半徑,在材料合成過程中,少量的Ni2+會遷移至Li層,從而破壞(003)晶面的有序程度,并導致(003)/(104)峰強比隨著Li+/Ni2+混排程度減小而增大,故NM?A樣品具有更低的Li+/Ni2+混排程度[8]。
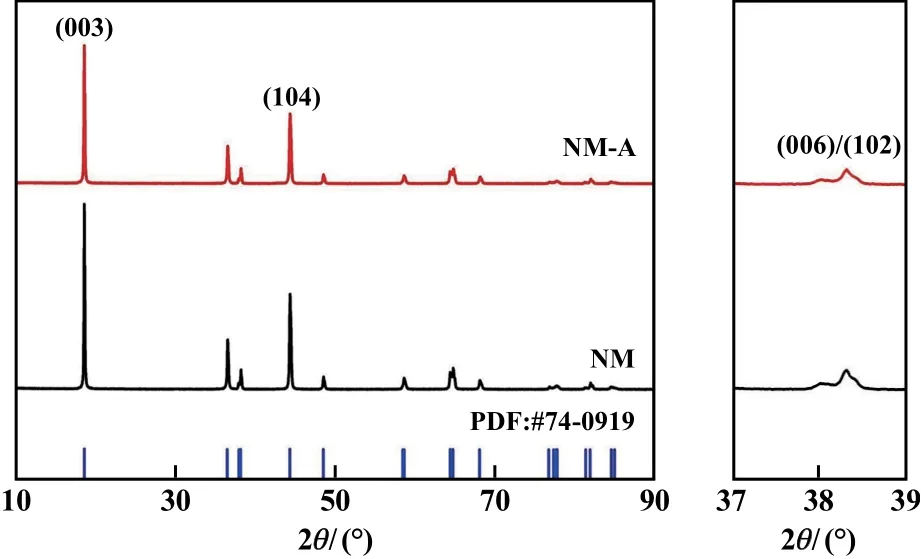
圖1 樣品XRD及局部放大圖
表1列出了2種材料的晶胞參數。可以看出,鋁摻雜會導致晶胞參數a減小、c增大,由此引起晶胞體積V略微減小。這是因為Al3+半徑(0.054 nm)略小于Ni3+半徑(0.056 nm),所以晶胞參數a和過渡金屬八面體TMO6層間距減小。另一方面,Al3+電負性(約1.513)比Ni3+電負性(約1.695)低,使得TM—O的結合更偏向于離子鍵,這會引起氧層之間的靜電斥力增強,進而導致層間距增大,故Al3+會導致晶胞參數c增大,有利于鋰離子在層狀結構中的擴散[9]。此外,這兩個樣品的c/a比值一樣且都大于4.9,表明鋁摻雜并不會降低層狀正極材料的結晶度。
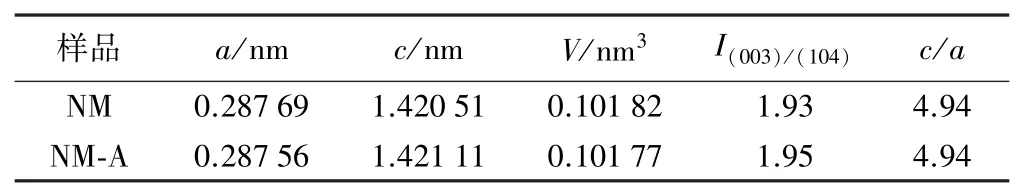
表1 樣品晶胞參數
2.2 微觀形貌及元素分布表征
圖2為原始樣品NM以及摻雜樣品NM?A的掃描電鏡(SEM)圖。2個樣品的二次顆粒均為球形,且尺寸大小不一,分布在8~12 μm范圍內。圖2(b)、(d)為正極材料顆粒的放大圖,可以看出,NM和NM?A的二次顆粒都是由立方體的一次顆粒聚集而成,但NM?A樣品表面的一次顆粒堆積更加緊密,孔徑數更少,這有利于抑制電解液進入顆粒內部。

圖2 樣品SEM圖
圖3為NM和NM?A樣品表面的EDS?Mapping能譜。可以看出,O、Ni和Mn元素都均勻地分布在2種正極材料顆粒上,而Al元素只出現在NM?A樣品表面,表明Al3+均勻地摻入到NM?A正極顆粒的表面。NM?A樣品的截面Mapping(圖3(c))進一步證明了Al3+成功地摻入到NM?A正極材料顆粒的體相中。

圖3 樣品EDS?Mapping圖
2.3 電化學性能測試
樣品首次充放電曲線見圖4。由圖4可知,測試溫度30℃時,NM?A樣品在電流密度0.1C、電壓區間2.7~4.3 V內的首次放電比容量和庫倫效率分別為202.8 mAh/g和83.6%,高于未摻雜的NM樣品(201.6 mAh/g,82.3%)。NM?A樣品充電平臺更低,而放電平臺更高,表明在充放電過程中,NM?A樣品極化程度更小。這歸因于NM?A樣品的低Li+/Ni2+混排程度和較大的層間距,從而在充放電過程中,Li+遷移時遇到的阻抗較小,遷移速率更快。但Al3+不像Ni3+一樣具有電化學活性,最終導致NM?A在放電末期容量衰減速率更快。
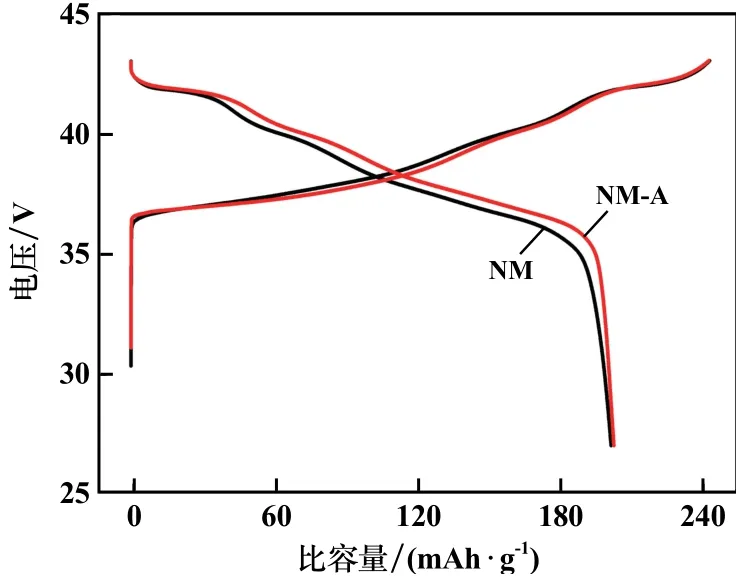
圖4 樣品首次充放電曲線
不同樣品倍率性能對比如圖5所示。Al3+的電化學惰性導致了NM?A樣品倍率性能降低,特別是在10C的高倍率條件下,NM?A樣品放電比容量僅為90.1 mAh/g,而未摻雜的NM樣品在同樣倍率下的放電比容量為107.9 mAh/g。
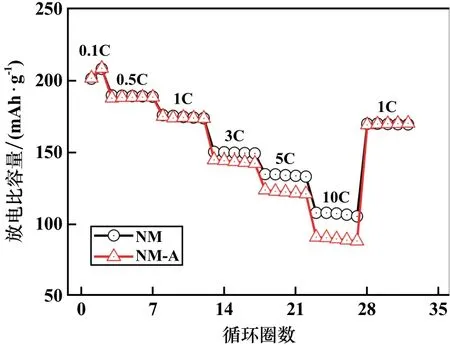
圖5 樣品倍率性能對比
圖6對比了NM和NM?A樣品在30℃、1C電流密度下的循環性能。鋁摻雜的NM?A樣品循環300圈后的容量保持率為71.2%,比未摻雜的NM樣品高出了11.3%。為了進一步探究鋁摻雜提高層狀無鈷LiNi0.95Mn0.05O2正極材料循環性能的機理,圖7采用微分容量曲線(dQ/dV)來分析樣品在循環過程中的相變。與其他高鎳層狀正極材料一樣,無鈷的NM材料在充放電過程中也會經歷多個相變:H1→M,M→H2,H2→H3,其中H表示六方相,M則表示單斜相,此相變過程還會導致材料發生晶胞參數和晶胞體積的變化。在充電過程中,正極材料的H2→H3相變階段會導致晶胞參數c急劇收縮,這種不均勻的體積變化會引起應力集中,進而誘發微裂紋的形成和擴展[10]。
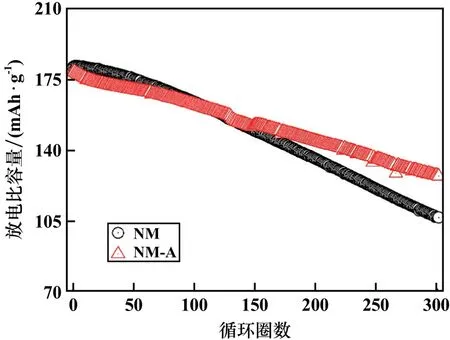
圖6 樣品循環性能對比
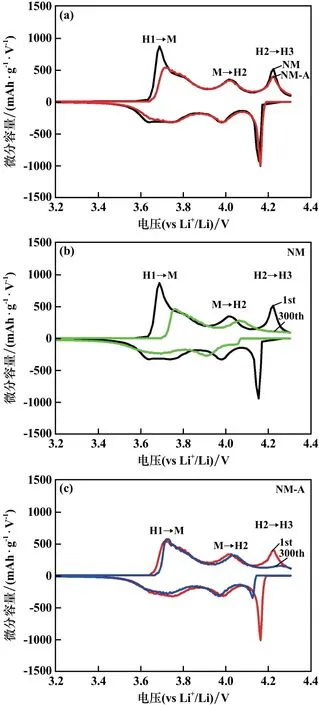
圖7 樣品微分容量曲線
如圖7所示,在首次循環過程中,NM?A的H2→H3氧化峰強度明顯低于NM樣品,表明NM?A的相變程度更小。這是因為NM?A樣品中存在鍵強更高的Al—O鍵(512 kJ/mol,Ni—O鍵強為391.6 kJ/mol),有利于穩定材料在充電態的層狀結構[11]。在經歷長循環后,NM?A樣品的H2→H3相變所對應的氧化峰強度衰減較慢,表明材料相變可逆性較好,這有利于緩解可逆容量的降低。考慮到Li+/Ni2+混排會促進材料在循環過程中由層狀結構向尖晶石相以及NiO巖鹽相轉變,因此鋁摻雜通過降低材料的Li+/Ni2+混排來提高H2→H3相變的可逆性。
圖8為循環后樣品的阻抗對比圖。其中位于高頻區的第1個半圓以及位于中頻區的第2個半圓分別代表表面膜阻抗(Rf)和電荷轉移阻抗(Rct)[12?13]。在經歷300圈的長循環后,NM?A正極的Rf和Rct分別為14.58 Ω和525.10 Ω,遠小于NM樣品的49.65 Ω和962.50 Ω,表明循環后NM正極材料的結構衰退程度更為嚴重,這與微分容量分析結果相對應。

圖8 300圈循環后樣品阻抗對比圖
3 結 論
1)鋁摻雜會引起材料晶胞參數的變化,并降低材料的Li+/Ni2+混排程度。SEM分析結果表明,Al均勻地摻雜到了正極材料二次顆粒的表面及體相中。
2)相對于NM樣品,1%鋁摻雜樣品擁有更高的首次放電比容量(202.8 mAh/g)和庫倫效率(83.6%)。但Al3+的非電化學活性會降低其倍率性能。
3)1%鋁摻雜可以提高LiNi0.95Mn0.05O2正極材料的長循環穩定性,這是因為鋁摻雜可以抑制材料在充電過程中的H2→H3相變程度并降低Li+/Ni2+混排,從而減小材料在長循環過程中可逆容量的衰減。
