基于機(jī)器學(xué)習(xí)的碳酸鹽微觀結(jié)構(gòu)與性質(zhì)
2022-08-29 10:52:14路貴民
華東理工大學(xué)學(xué)報(自然科學(xué)版) 2022年4期
關(guān)鍵詞:方法
楊 博, 路貴民
(華東理工大學(xué)國家鹽湖資源綜合利用工程技術(shù)研究中心,上海 200237)
熔鹽作為一種傳蓄熱介質(zhì),廣泛應(yīng)用于聚焦式光熱發(fā)電(Concentrating Solar Power, CSP)中[1-2]。在美國國家能源局提出的第3 代光熱發(fā)電技術(shù)的路線圖中,將采用超臨界二氧化碳-布雷頓循環(huán)系統(tǒng)代替現(xiàn)有的蒸汽-朗肯循環(huán)系統(tǒng)進(jìn)行發(fā)電[3]。這項新的技術(shù)要求傳蓄熱介質(zhì)的工作溫度為520~720 ℃,熱穩(wěn)定性優(yōu)異且腐蝕性相對較小的碳酸鹽[3-4]可以滿足這一要求。研究碳酸鹽的微觀結(jié)構(gòu)以及微觀性質(zhì)可以幫助提高基于碳酸鹽的傳蓄熱介質(zhì)的性能,但是,開展高溫熔鹽結(jié)構(gòu)信息的實驗研究比較困難,高溫實驗也容易造成數(shù)據(jù)波動和誤差。
隨著計算機(jī)技術(shù)的進(jìn)步,計算機(jī)模擬發(fā)展成為一項成熟的分子模擬技術(shù)。基于密度泛函理論(Density Functional Theory, DFT)的量子力學(xué)計算以及經(jīng)典分子動力學(xué)(Classical Molecular Dynamics,CMD)模擬廣泛應(yīng)用于熔鹽的分子模擬領(lǐng)域[5-12]。Koishi 等[13]在Li2CO3-K2CO3的CMD 模擬計算中采用了基于Tosi-Fumi 公式的勢函數(shù)進(jìn)行運算,但是他將視作1 個以C 為中心,3 個O 呈等邊三角形分布的固定分子,忽略了的分子內(nèi)作用。Costa[14]在Li2CO3-K2CO3的CMD 模擬中采用FC 極化模型(Fluctuating Charge Model),將的分子內(nèi)作用引入模擬體系中。Du 等[15]利用包含庫侖項的BornMayer勢函數(shù)模擬了Na2CO3的結(jié)構(gòu)與性質(zhì),計算結(jié)果表明模擬計算的為一個金字塔型的立體結(jié)構(gòu)。Desmaele 等[16]采用CMD方法計算碳酸鹽的結(jié)構(gòu)與性質(zhì),用3 個經(jīng)驗公式分別描述的C-O 相互作用、O-O 相互作用以及與陽離子的相互作用,經(jīng)驗公式的計算參數(shù)由基于DFT 的AIMD(ab initio Molecular Dynamics)計算優(yōu)化,計算結(jié)果表明熔融狀態(tài)下碳酸鹽的陰陽離子之間處于相互牽扯的狀態(tài),陽離子包裹陰離子并且將其引入第2 配位層,同時陰離子的1~2 個O 原子包裹著陽離子;還發(fā)現(xiàn)包裹陽離子的O 原子數(shù)量不受陽離子種類和大小的影響。Kiyobayashi等[17]計算了A2CO3(A 為Li、Na、K、 Rb、Cs)的離子電導(dǎo)率,用Born-Mayer-Huggins勢函數(shù)描述離子之間作用,并用3 個經(jīng)驗公式描述的鍵角。Roest 等[18]在對碳酸熔鹽燃料電池的帶電界面進(jìn)行CMD 模擬時同樣采用Born-Mayer-Huggins 勢函數(shù)描述離子之間作用。
勢函數(shù)和經(jīng)驗參數(shù)是CMD 模擬的基礎(chǔ),勢函數(shù)及經(jīng)驗參數(shù)的選擇是否合理直接影響計算結(jié)果的準(zhǔn)確度,通常勢函數(shù)以及參數(shù)通過實驗結(jié)果或者量子力學(xué)計算進(jìn)行優(yōu)化。本文采用基于DFT 理論的第一性原理計算(First-principle Molecular Dynamics, FPMD)、機(jī)器學(xué)習(xí)(Machine Learning, ML)和CMD 方法,建立深度勢分子動力學(xué)模擬方法(Deep Potential Molecular Dynamics, DPMD)。在DPMD 方法中,ML 將DFT 的結(jié)果擬合為CMD 可以識別的深度勢(Deep Potential,DP),而不是通過DFT 優(yōu)化CMD的勢函數(shù)。本文對K2CO3和Na2CO3進(jìn)行DPMD 模擬,計算K2CO3和Na2CO3的結(jié)構(gòu)信息及其部分物理性質(zhì),探究DPMD是否可以應(yīng)用于碳酸鹽體系。
1 計算方法
1.1 第一性原理計算
采用 VASP.5.4.4(Vienna ab-initio Simulation Package)軟件包進(jìn)行第一性原理計算。PBE (Perdew-Burke-Ernzerhof)函數(shù)用于交換和關(guān)聯(lián)計算[19],PAW(Projector-Augmented Wave)用于描述原子核和價電子的相互作用[20-21]。氧的價電子構(gòu)型選擇為2s22p4,碳的價電子構(gòu)型選擇為2s22p2,鉀的價電子構(gòu)型選擇為3s23p64s1。根據(jù)VASP 參數(shù)手冊的推薦,截斷能設(shè)置為560 eV,參數(shù)K 點設(shè)置為1×1×1,模擬過程中的溫度控制采用Nose-Hoover 恒溫器方法。
第一性原理的計算分為3 步。在溫度2 000 K 下運行6 000 個時間步(6 ps)快速獲取模型的合理構(gòu)型,然后以179 K/ps 的冷卻速率降溫至目標(biāo)溫度1 180、1 230、1 280 K,最后在目標(biāo)溫度下運行20 000 個時間步(20 ps)。
1.2 擬合深度勢 (DP Train)
DeepMD-kit.1.2 軟件包用于構(gòu)建深度勢函數(shù)模型[22-23]。系統(tǒng)的能量定義為每個原子能量的加和,如式(1)所示:

其中:Ei是以原子i為球心,截斷半徑(rc)為半徑的球形空間的能量,Ei定義如式(2)所示:
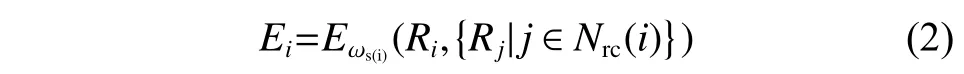
其中:Ri={xi,yi,zi} ,表示原子i的坐標(biāo);Rj={xj,yj,zj} ,表示原子j的坐標(biāo);Nrc(i)是模擬過程中原子i的臨近列表;s(i)表示原子i的化學(xué)類別。Ei對Ri的依賴關(guān)系通過深度神經(jīng)網(wǎng)絡(luò)(Deep Neural Network, DNN)參數(shù)化,DNN 的參數(shù)ω由機(jī)器學(xué)習(xí)過程控制,采用Adam method 方法優(yōu)化[24]。
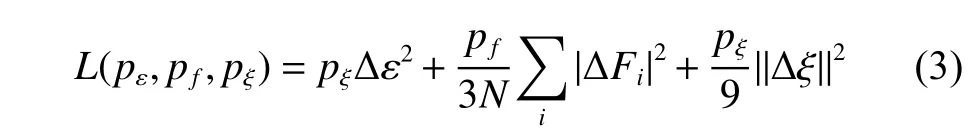
其中:L表示損失函數(shù);pε、pf、pξ是可調(diào)節(jié)因子;N是原子的總數(shù)目; Δ ε 代表機(jī)器學(xué)習(xí)預(yù)測數(shù)據(jù)與對應(yīng)的第一性計算結(jié)果的能量的差值; ΔFi代表機(jī)器學(xué)習(xí)預(yù)測數(shù)據(jù)與對應(yīng)的第一性計算結(jié)果的受力的差值; Δ ξ 是預(yù)測數(shù)據(jù)與第一性計算結(jié)果張量除以N的差值。
DeepMD-kit.1.2 中,Zhang 等[25]提出一個靈活的描述符,通過將坐標(biāo)轉(zhuǎn)化為矢量的方法來保持體系的對稱性,解決了模擬體系的原子坐標(biāo)無法直接應(yīng)用于機(jī)器學(xué)習(xí)過程的問題,許多計算工作驗證了其可靠性[23,26-27]。根據(jù)式(4),體系的原子坐標(biāo)轉(zhuǎn)變?yōu)榘瑥较蛐畔⒑徒嵌刃畔⒌膹V義坐標(biāo):
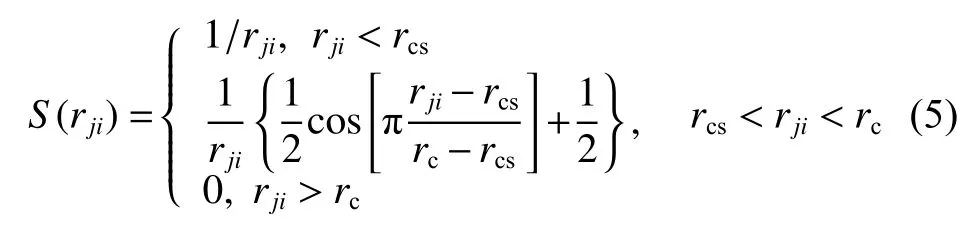
其中:rji為原子j與原子i之間的距離;rcs表示截斷半徑的平滑參數(shù)。
使用DNN 將徑向信息映射到矩陣gi1和gi2上,最終,根據(jù)式(6),廣義坐標(biāo)重新編碼為特征矩陣Di。
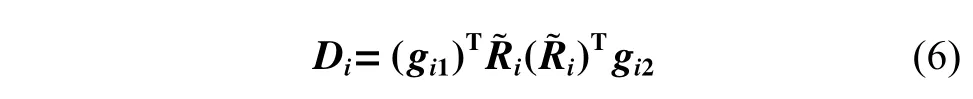
第一性原理的計算結(jié)果使用python 代碼處理,作為機(jī)器學(xué)習(xí)的輸入數(shù)據(jù)集。機(jī)器學(xué)習(xí)的過程中,第一性原理計算的前15 000 個時間步(15 ps)的結(jié)果用于擬合深度勢,后5 000 個時間步(5 ps)的計算結(jié)果用于測試深度勢的精度。嵌入神經(jīng)網(wǎng)絡(luò)設(shè)置3 個隱藏層,隱藏層大小分別為25、50、100,擬合神經(jīng)網(wǎng)絡(luò)大小設(shè)置為240、240、240,深度學(xué)習(xí)過程共持續(xù)1 000 000 步。
1.3 經(jīng)典分子動力學(xué)模擬
CMD 的計算使用Lammps(Large-scale Atomic/Molecular Massively Parallel Simulator)軟件包完成,采用1.2 節(jié)中的深度勢代替勢函數(shù)公式進(jìn)行運算。計算中采用周期性邊界條件(Periodic-Boundary-Condition,PBC)消除表面效應(yīng),保證體系粒子數(shù)目恒定,采用Verlet 算法求解牛頓方程,設(shè)置時間步長=1 ps[28]。
學(xué)生雖然深知“數(shù)學(xué)解題就是由未知向已知不斷轉(zhuǎn)化的過程”,但考試時卻又常常受制于“轉(zhuǎn)化目標(biāo)不明”和“調(diào)控受阻思維能力不強(qiáng)”兩大因素,導(dǎo)致無謂失分.因此,從教“怎樣想”入手,深入挖掘中考試題解題思路的生成過程,培養(yǎng)學(xué)生良好的思維習(xí)慣,對提升轉(zhuǎn)化能力必大有裨益.
Lammps 模擬計算中,計算開始時原子會獲得一個遵循高斯分布的隨機(jī)初速度,性質(zhì)計算均在等溫等壓系綜(NPT)或者正則系綜(NVT)下進(jìn)行,溫度控制使用Nose-Hoover 恒溫器方法和Barostat 方法[29-30]。
徑向分布函數(shù)gαβ(r) 和配位數(shù)Nαβ是表征熔鹽微觀結(jié)構(gòu)的常用方法,其計算公式如式(7)和式(8)所示:
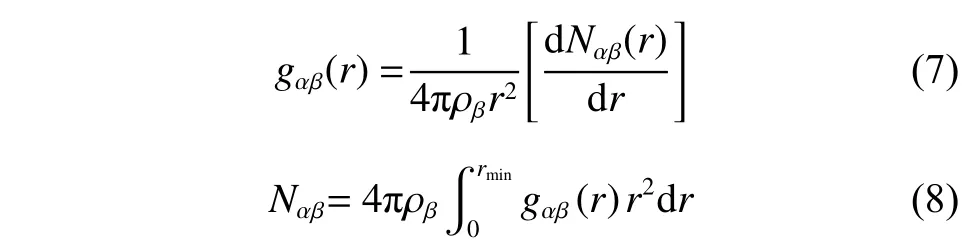
其中: ρβ是β原子的數(shù)密度;r表示兩個原子之間的距離;Nαβ(r) 是式(1)中提到的微小空間內(nèi)β原子的平均數(shù)目;rmin是徑向分布函數(shù)(RDF)第一峰峰谷的位置。
密度計算根據(jù)式(9)在NPT 系綜下進(jìn)行:
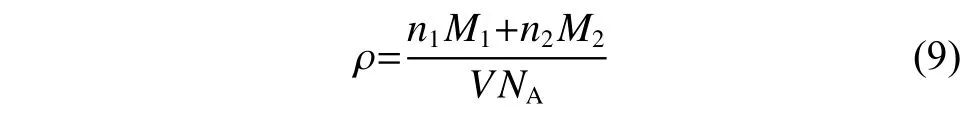
其中:n是原子的數(shù)目;M是摩爾質(zhì)量;V是體系平衡體積;NA是阿伏伽德羅常數(shù)。
比熱容(Cp)用于描述熔鹽吸放熱能力的強(qiáng)弱,在模擬計算密度時,可以通過統(tǒng)計體系的焓值隨溫度的變化情況來計算,計算公式如式(10)所示:
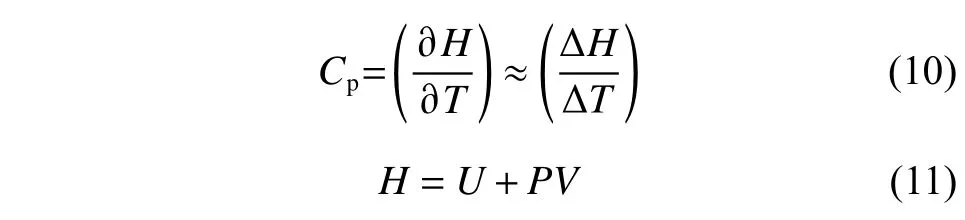
其中:H為體系的焓值;P為壓力;U為系統(tǒng)內(nèi)能,包括動能與勢能。
熱導(dǎo)率采用逆非平衡分子動力學(xué)方法(Reverse Nonequilibrium Molecular Dynamics Method,rNEMD)[31-32]計算。將體系沿z軸方向擴(kuò)展1 倍然后等分為n層,將第1 層動能最大的原子與(n/2+1)層動能最小的原子每隔一段時間進(jìn)行動能交換,等待體系達(dá)到動態(tài)平衡后統(tǒng)計體系中動能交換的大小以及監(jiān)測體系的溫度梯度,可通過式(12)計算熱導(dǎo)率:

黏度 ( η) 采 用EMD 方法(Equilibrium Molecular Dynamics Method)進(jìn)行計算,EMD 方法基于Green-Kubo 公式,通過計算應(yīng)力張量自相關(guān)系數(shù)(Stress Tensor Auto-correlation Functions,SACF)得到體系的黏度。Green-Kubo 公式如下:
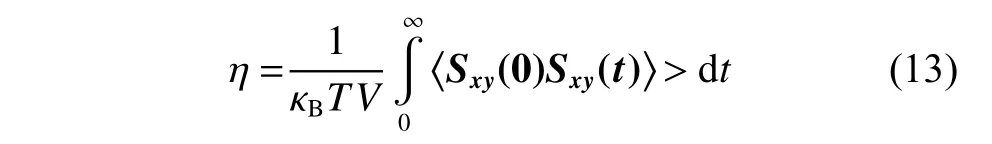
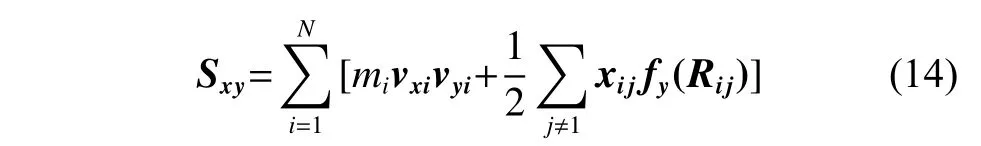
其中:mi為原子i的質(zhì)量;vxi和vyi分別為原子i在x和y方向的速度分量;fy(Rij)是原子j對原子i在y方向的作用力;Rij表示原子i、j的適量距離;xij是Rij在x方向的分量。
2 結(jié)果與討論
2.1 機(jī)器學(xué)習(xí)的誤差
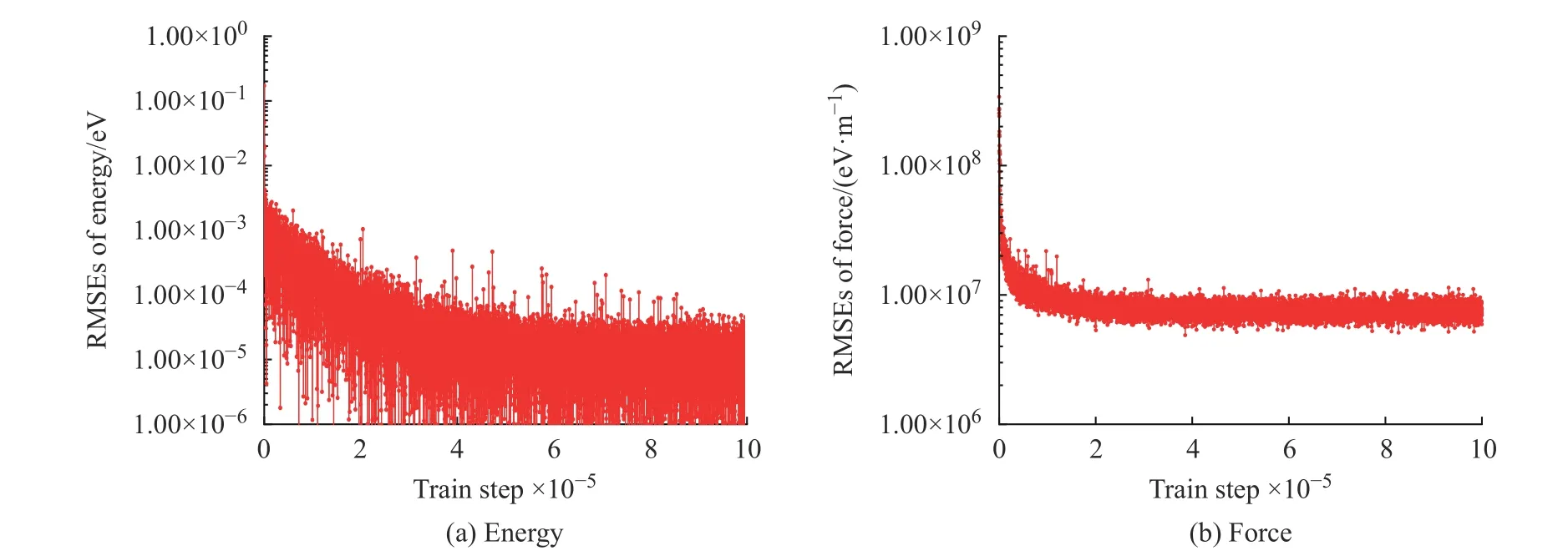
圖1 學(xué)習(xí)過程中K2CO3 能量和受力的均方根誤差Fig. 1 EMSEs of energy and force of K2CO3 during learning process
學(xué)習(xí)過程中K2CO3能量和受力的均方根誤差(Root Mean Square Error, RMSEs)如圖1 所示,圖中數(shù)據(jù)說明6×105~10×105個步長的訓(xùn)練滿足擬合勢函數(shù)模型的要求,隨著學(xué)習(xí)時間的增加,能量的均方根誤差在1.00×10-4eV 以下,受力的均方根誤差在1.00×107eV/m 附近。模型的準(zhǔn)確率通過執(zhí)行擬合驗證數(shù)據(jù)與擬合數(shù)據(jù)的對比測試進(jìn)行評估,K2CO3機(jī)器學(xué)習(xí)的預(yù)測結(jié)果與相對應(yīng)的DFT 計算結(jié)果的對比如圖2所示,預(yù)測結(jié)果與DFT計算值吻合度較好。DeepMDkit.1.2 軟件包執(zhí)行對比測試后顯示K2CO3能量和受力的均方根誤差非常低,分別為8.62×10-4eV 和4.67×108eV/m,表示勢函數(shù)模型的精度與DFT 精度接近。圖3 示出了學(xué)習(xí)過程中Na2CO3能量與受力的均方根誤差。圖4 示出了Na2CO3機(jī)器學(xué)習(xí)的預(yù)測結(jié)果與DFT 計算值的對比。由圖可知,隨著學(xué)習(xí)時間的增加,Na2CO3能量的均方根誤差在1.00×10-4eV 以下,受力的均方根誤差在1.00×109eV/m 以下,執(zhí)行對比測試后能量與受力的均方根誤差分別為1.19×10-3eV 和5.31×108eV/m。
2.2 結(jié)構(gòu)分析
圖5 示出了不同溫度下采用DPMD 和DFT方法計算所得的K2CO3徑向分布函數(shù)曲線的對比,二者的計算結(jié)果吻合度較好,表明DPMD 方法可以準(zhǔn)確描述K2CO3熔融狀態(tài)下離子之間的相互作用。
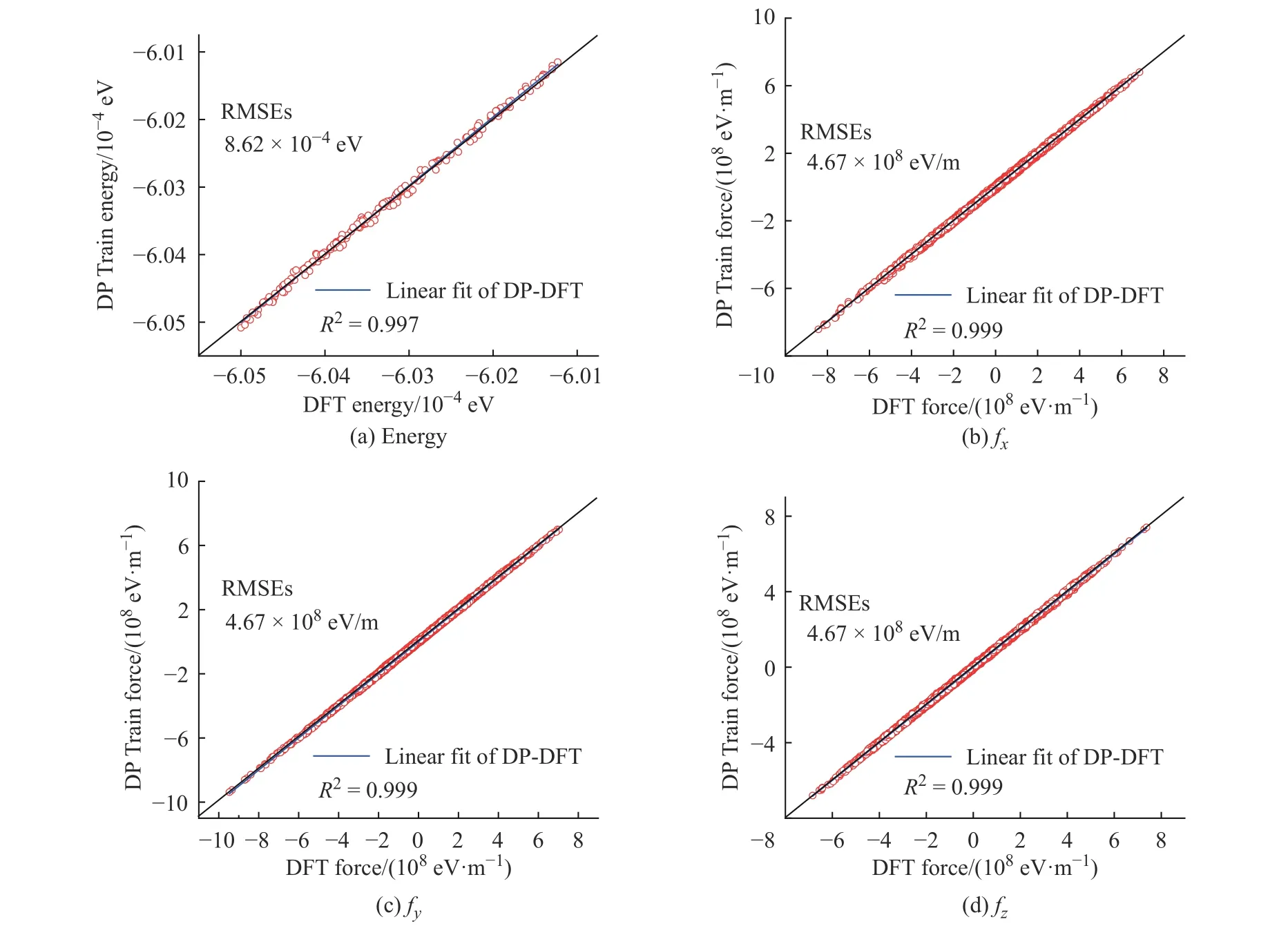
圖2 K2CO3 機(jī)器學(xué)習(xí)的預(yù)測結(jié)果與DFT 計算結(jié)果的對比Fig. 2 Comparsion of ML prediction result and DFT calculation result of K2CO3
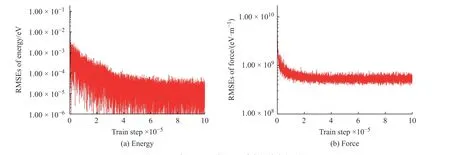
圖3 學(xué)習(xí)過程中Na2CO3 能量和受力的均方根誤差Fig. 3 RMSEs of energy and force of Na2CO3 during learning process
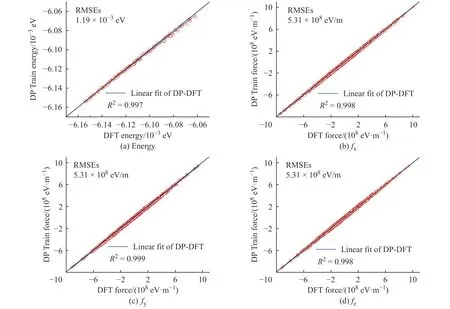
圖4 Na2CO3 機(jī)器學(xué)習(xí)的預(yù)測結(jié)果與DFT 計算結(jié)果的對比Fig. 4 Comparsion of ML prediction result and DFT calculation result of Na2CO3
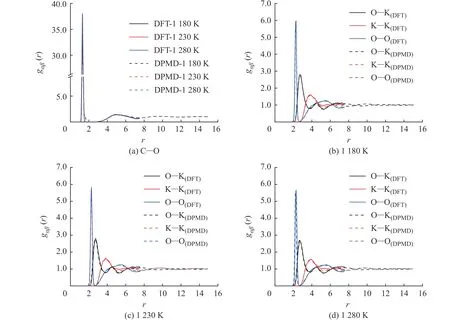
圖5 不同溫度下K2CO3 徑向分布函數(shù)的DPMD 與DFT 計算結(jié)果對比Fig. 5 Comparsion of radial distribution functions of K2CO3 derived from DPMD and DFT calculation at different temperatures
圖6 示出了不同溫度下采用DPMD 方法計算的K2CO3部分離子對的徑向分布函數(shù)曲線。在熔融狀態(tài)下,所有離子對的徑向分布函數(shù)曲線都具有相同的特點:每一條曲線都具有非常高的第一峰,在第一峰后的第二峰高度迅速降低,隨著距離的增大,徑向分布函數(shù)曲線逐漸趨近于1.0,這與離子液體“近程有序、長程無序”的特點相對應(yīng)。第一峰的峰形可以解讀出離子對作用的強(qiáng)弱,高而尖的峰代表著排列緊湊有序,矮而寬的峰則代表松散無序。圖6(a)中第一峰的峰高隨著溫度升高而降低,表明隨著溫度的增加C-O 的相互作用減弱,說明隨著溫度的升高而略有松散。O-K、K-K 和O-O 的徑向分布函數(shù)曲線分別如圖6(b)、6(c)、6(d)所示,相比于C-O 曲線,O-K、K-K 和O-O 曲線第一峰的峰高隨溫度升高有一定程度的降低,O-K 第一峰的峰位置隨著溫度升高略微向右邊移動,表明K+與的作用距離在增大。各個離子對的第二峰峰值減弱,峰形變寬,O-O 對的第二峰呈現(xiàn)平臺狀,而第三峰基本觀察不到,說明粒子的不規(guī)則性增大,因此可以判斷形成了團(tuán)簇。
圖7 示出了采用DPMD 和DFT 方法計算所得的Na2CO3徑向分布函數(shù)曲線的對比,二者的計算結(jié)果吻合度較好。圖8 示出了DPMD 方法計算所得的Na2CO3部分離子對的徑向分布函數(shù)曲線,所有離子對均保持“近程有序、長程無序”的特點,C-O、O-Na、Na-Na 和O-O 離子對曲線的第一峰的峰高均隨著溫度的升高而降低,說明離子對之間的相互作用在逐漸減弱。圖8(a)中C-O 離子對在各個溫度點下的徑向分布函數(shù)曲線的分布趨勢高度相似,表明隨著溫度升高結(jié)構(gòu)保持穩(wěn)定。O-Na、Na-Na 和O-O 離子對的徑向分布函數(shù)曲線分別如圖8(b)、8(c)、8(d)所示,在1 230 K 和1 260 K 下,3 個離子對的徑向分布函數(shù)曲線第一峰后的曲線歸一化趨勢更加明顯,表明溫度升高以后Na2CO3體系中陰陽離子的不規(guī)則性增大。
2.3 性質(zhì)分析
2.3.1 密度 密度是碳酸鹽工業(yè)應(yīng)用的基礎(chǔ)性質(zhì)之一,根據(jù)式(9)模擬計算了K2CO3和Na2CO3的密度,計算結(jié)果如圖9 所示。K2CO3模擬值比文獻(xiàn)結(jié)果[33]高約5.0%,Na2CO3模擬值比文獻(xiàn)結(jié)果[33]高約5.6%。隨著溫度升高,逐漸變得松散以及K、Na 和O 的距離變大,導(dǎo)致K2CO3和Na2CO3的摩爾體積增大,因此密度與溫度呈負(fù)相關(guān)。根據(jù)計算值擬合出K2CO3和Na2CO3的密度計算公式分別如式(15)、式(16)所示。
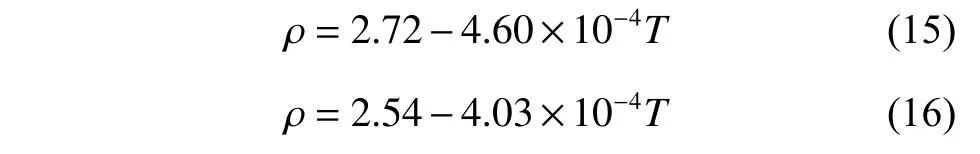
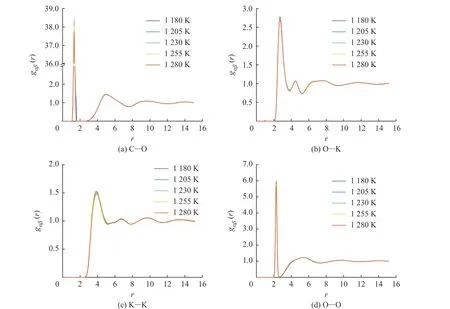
圖6 DPMD 方法計算所得的K2CO3 徑向分布函數(shù)Fig. 6 Radial distribution functions of K2CO3 calculated by DPMD method
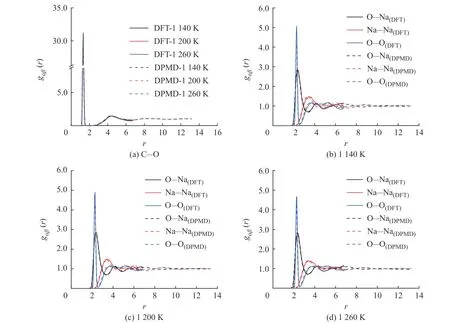
圖7 不同溫度下Na2CO3 徑向分布函數(shù)的DPMD 與DFT 計算結(jié)果對比Fig. 7 Comparsion of radial distribution functions of Na2CO3 derived from DPMD and DFT calculation at different temperatures
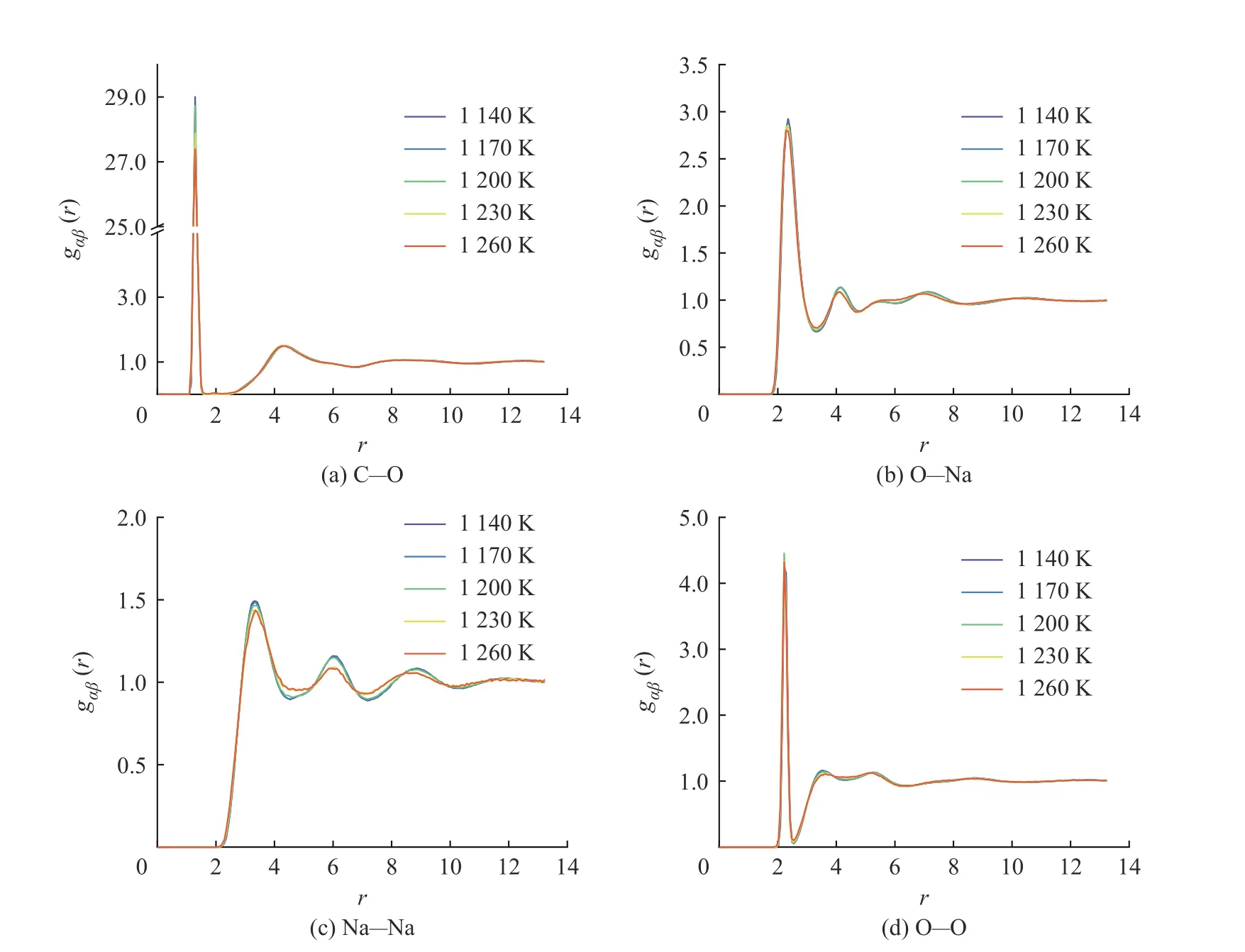
圖8 DPMD 方法計算所得的Na2CO3 徑向分布函數(shù)Fig. 8 Radial distribution functions of Na2CO3 calculated by DPMD method
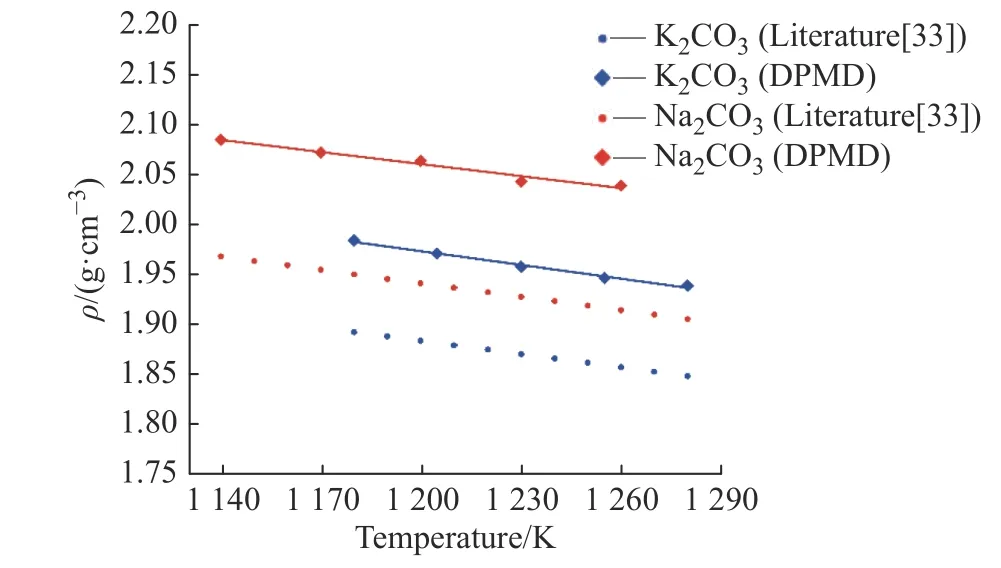
圖9 不同溫度下K2CO3 和Na2CO3 密度的文獻(xiàn)值與模擬值對比Fig. 9 Comparison of literature and simulated density of K2CO3 and Na2CO3 at different temperatures
2.3.2 比熱容 根據(jù)公式(10),K2CO3和Na2CO3的溫度-焓曲線如圖10 所示,曲線斜率即溫度區(qū)間內(nèi)的比熱容,該比熱容為溫度區(qū)間內(nèi)的平均比熱容。K2CO3和Na2CO3的平均比熱容與文獻(xiàn)值[33]統(tǒng)計于表1 中,計算偏差分別為3.3%和6.0%。
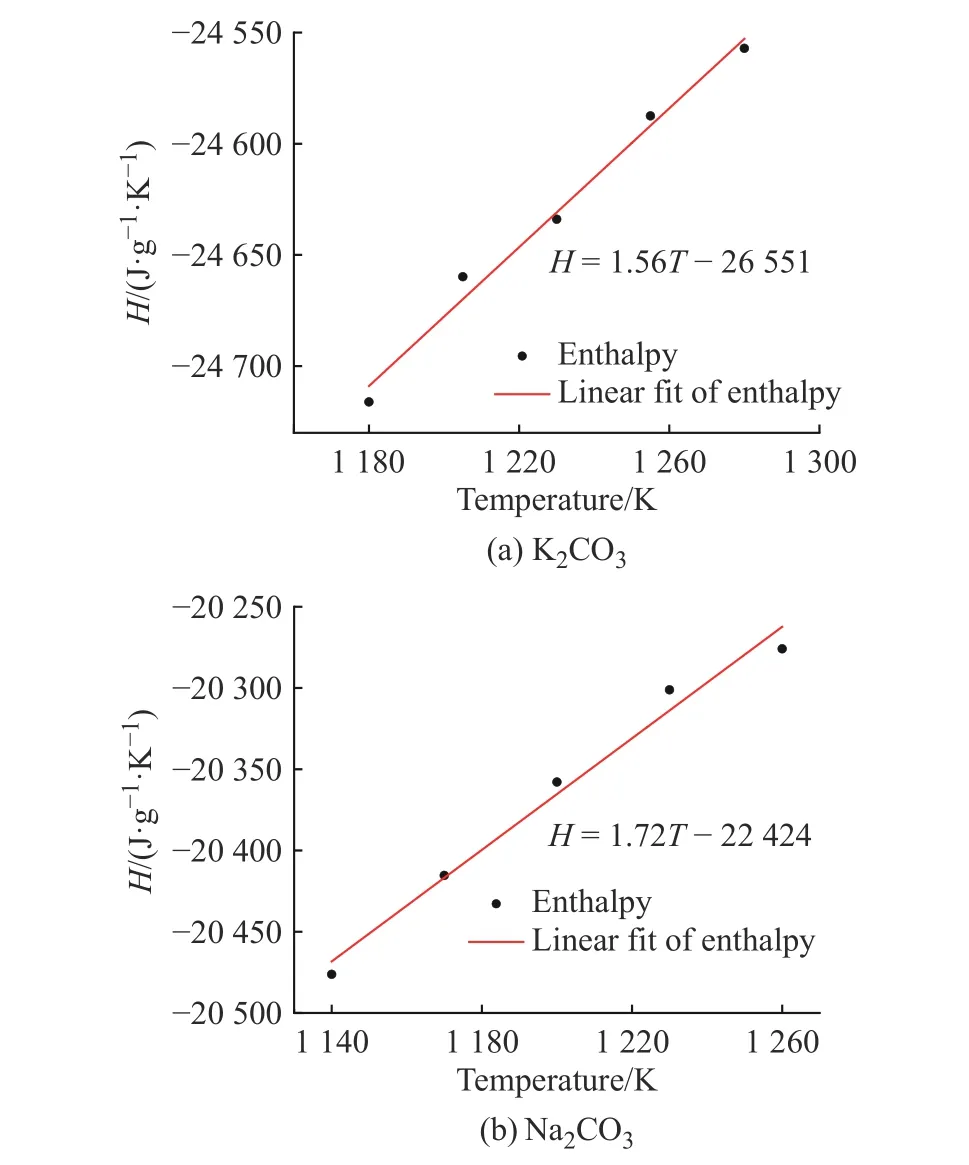
圖10 K2CO3 和Na2CO3 計算中焓隨溫度的變化Fig. 10 Changes of enthalpy of K2CO3 and Na2CO3 with temperature
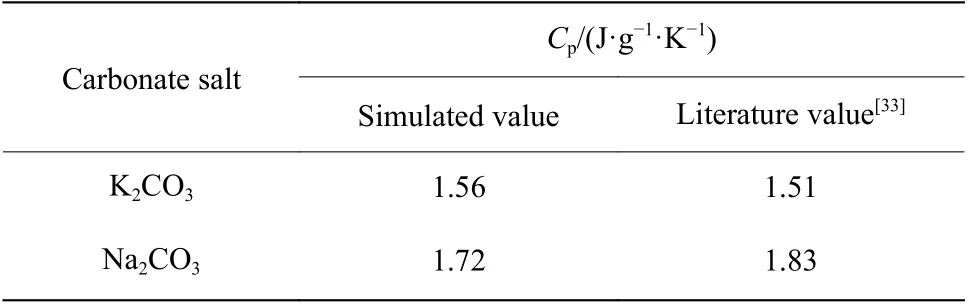
表1 K2CO3 和Na2CO3 比熱容的模擬值與文獻(xiàn)值對比Table 1 Comparison of simulated and literature values of the Cp of K2CO3 and Na2CO3
2.3.3 熱導(dǎo)率 圖11 示出了K2CO3和Na2CO3熱導(dǎo)率的文獻(xiàn)值[34]與模擬值對比,其熱導(dǎo)率計算偏差分別約為8.0%和3.5%。從計算點擬合的趨勢線發(fā)現(xiàn),K2CO3和Na2CO3的熱導(dǎo)率隨著溫度的升高而降低,在熱量傳遞過程中,粒子只需要在局域范圍內(nèi)振蕩,且只需與相鄰的粒子發(fā)生作用而不涉及長程運動,隨著溫度升高,體系的摩爾體積增大,離子間距增大,影響了體系通過離子振動傳遞能量的能力,導(dǎo)致隨著溫度升高熱導(dǎo)率變小。
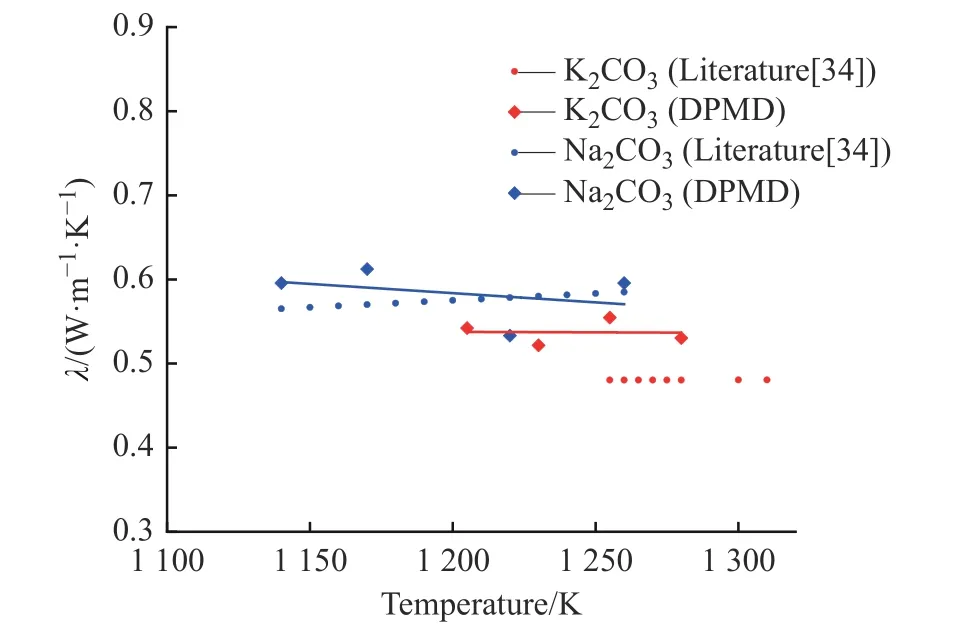
圖11 K2CO3 和Na2CO3 熱導(dǎo)率的文獻(xiàn)值與模擬值對比Fig. 11 Comparison of literature and simulated thermal conductivity of K2CO3 and Na2CO3
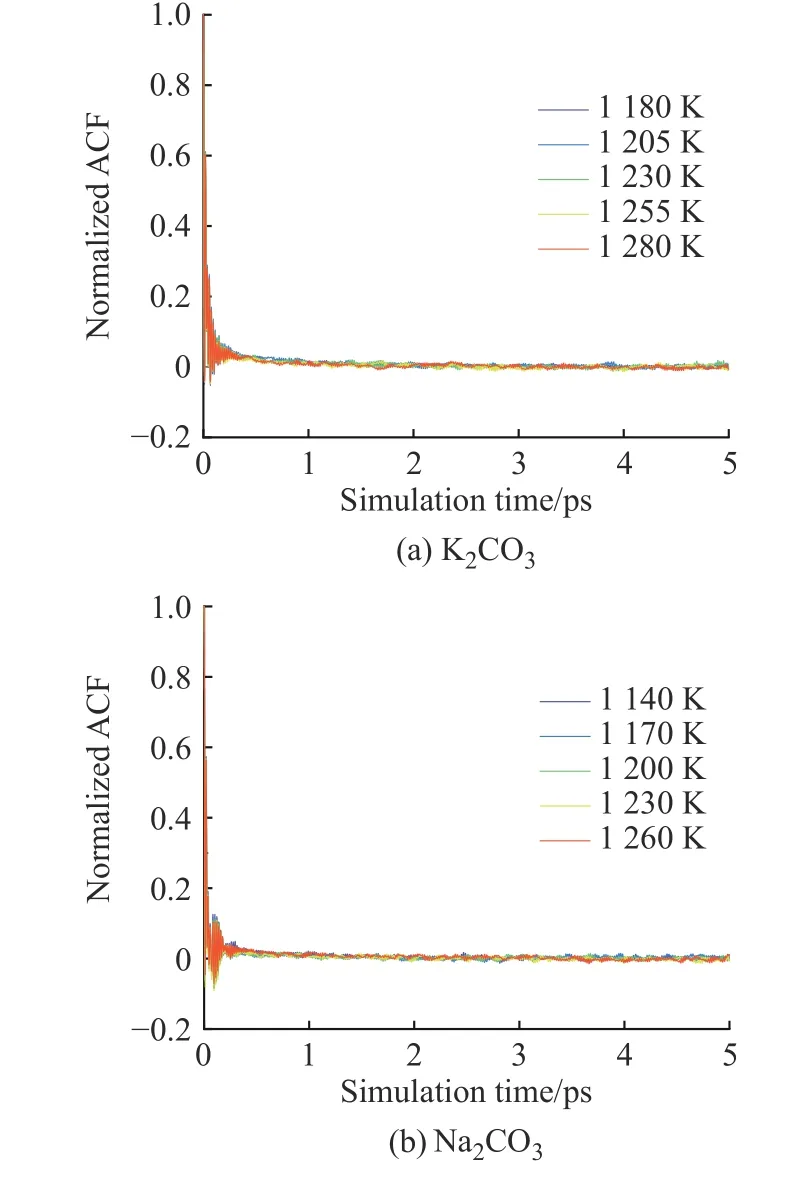
圖12 K2CO3 和Na2CO3 歸一化張量自相關(guān)函數(shù)Fig. 12 Normalized stress tensor auto-correlation function of K2CO3 and Na2CO3
2.3.4 黏度 K2CO3和Na2CO3的黏度采用平衡分子動力學(xué)模擬方法計算。圖12(a)、12(b)分別示出了用Green-Kubo 方法計算K2CO3和Na2CO3的歸一化張量自相關(guān)函數(shù)(ACF)。在模擬時長為5 ps 時,ACF趨近于0 并保持穩(wěn)定,說明5 ps 的計算時長完全可以滿足EMD 方法計算黏度的要求。圖13 示出了K2CO3和Na2CO3黏度的文獻(xiàn)值[33]與模擬值的對比。由圖可知,所得的計算黏度隨著溫度的升高而降低,并且在溫度較高的區(qū)域與文獻(xiàn)值比較接近。
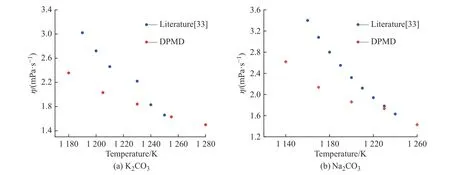
圖13 K2CO3 和Na2CO3 黏度的文獻(xiàn)值與模擬值對比Fig. 13 Comparison of literature and simulated viscosity of K2CO3 and Na2CO3
3 結(jié) 論
(1)采用基于機(jī)器學(xué)習(xí)的分子動力學(xué)模擬方法,對碳酸熔鹽的微觀結(jié)構(gòu)進(jìn)行了研究。利用第一性原理的計算結(jié)果作為訓(xùn)練深度勢的數(shù)據(jù)集,通過DNN訓(xùn)練深度勢函數(shù),機(jī)器學(xué)習(xí)為快速獲得準(zhǔn)確的勢函數(shù)提供了可靠的方法。
(2)K2CO3的能量和受力的均方根誤差分別為8.62×10-4eV和4.67×108eV/m,Na2CO3的能量與受力的均方根誤差分別為1.19×10-3eV 和5.31×108eV/m。在結(jié)構(gòu)計算中,DPMD 與DFT 的計算結(jié)果吻合較好,說明DPMD 對DFT 有良好的繼承性。DPMD 計算結(jié)果表明碳酸根為一個整體存在于碳酸鹽中,升高溫度會造成碳酸根團(tuán)簇的松散但不會破壞其正三角形結(jié)構(gòu),同時升高溫度會造成陽離子與碳酸根的間距增大,使整個體系的體積增大。在結(jié)構(gòu)計算中,對比K2CO3和Na2CO3性質(zhì)的模擬值與文獻(xiàn)值后發(fā)現(xiàn)數(shù)據(jù)較為接近,K2CO3的密度、比熱容和熱導(dǎo)率的計算誤差分別約為5.0%、3.3%和8.0%;Na2CO3的密度、比熱容和熱導(dǎo)率的計算誤差分別約為5.6%、6.0%和3.5%。
(3)本文的計算證明DPMD 在液態(tài)熔鹽中的應(yīng)用是可靠的,深度勢函數(shù)不僅繼承了DFT 的精確性,而且將計算范圍從DFT 的溫度點擴(kuò)展到了CMD 的溫度范圍。這意味著DPMD 擺脫了經(jīng)驗參數(shù)的束縛,可以實現(xiàn)高精度、高效率、大尺寸和長時間的模擬。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
