胰高血糖素樣肽-1受體激動劑和鈉葡萄糖共轉運體-2抑制劑的中樞神經系統靶點
2022-08-03 07:10:48董梅園許程琳李彥彥金建蘭周里鋼
西南醫科大學學報 2022年4期
文 松,董梅園,許程琳,李彥彥,金建蘭,龔 敏,周里鋼,2
1.上海市浦東醫院(復旦大學附屬浦東醫院)內分泌科(上海 201399);2.上海市血管病變調控與重塑重點實驗室(上海 201399)
肥胖癥和2 型糖尿病是復雜的慢性疾病,兩者的發病和治療又是密不可分的。肥胖患病率的增加,在一定程度上是由于全球食品供應的增加,為人們提供了無處不在的美味、高能量食品。高能量美食往往通過激活大腦食欲控制回路,通過增強食物尋求、進食動機和進食愉悅感來驅動貪食癥和體重增加。行為療法(飲食和生活方式的改變)是肥胖和2型糖尿病的首選治療方法,但它往往無法實現有意義的減肥。此外,通過這種方式減肥的人會通過生物調節逐漸恢復體重。從生物學的角度來講,人類為了維持正常的體內生理功能和活動,需要從食物中獲取能量。食物的攝入可以受到饑餓、進食頻率和應激壓力等條件的影響。病態的貪食和肥胖反映了有機體無法有效地維持能量平衡[1]。雖然越來越多的科研證據提示2 型糖尿病的發病和許多基因的變異(genetic variations)、基因拷貝數目的變異(copy number variations),以及基因修飾—DNA 甲基化和組蛋白修飾等相關[2],但肥胖、代謝綜合征仍是2 型糖尿病形成的最主要的原因,同時肥胖的治療,也是在沒有明確2 型糖尿病的具體基因病因之前,最直接和最有效的臨床治療方式[3-4]。在生物體中,能量平衡相關的生理和行為由神經和體液系統所控制。由于臨床上治療2 型糖尿病和肥胖癥的困難性,闡明中樞神經系統(central nervous system,CNS)能量平衡機制和確定CNS 中的治療關鍵靶點是目前肥胖及2型糖尿病治療的熱點。本文旨在總結:①作用在CNS上控制飲食和肥胖的關鍵位點;②目前臨床使用的最新和有效的藥物,GLP-1RA和SGLT-2i是如何通過中樞神經系統的能量平衡機制和控制交感神經的機制改善肥胖及慢性心衰的。本文通過討論GLP-1RA 和SGLT-2i治療引起中樞神經系統回路的激活,展示其有望成為肥胖和2型糖尿病的治療藥物。
1 中樞神經系統對能量和代謝的調節
能量代謝的調節涉及有機體的能量穩態和高級神經活動如獎賞行為的調節機制。其包括神經和體液內分泌、腸道、肝臟、棕色脂肪組織(brown adipose tissue,BAT)以及骨骼肌肉,正是通過這些系統的相互作用,機體能夠有效控制進食,維持內環境的穩態,故本文將從這幾方面探討能量代謝的調節。而其中下丘腦的眾多核團是主要的能量穩態調節中樞,由內側區和外側區組成。內側區由前、中、后3組神經元組成。弓狀核(arcuate nucleus,ARC)、腹內側核(ventromedial hypothalamic nucleus,VMN)、背內側核(dorsomedial hypothalamic nucleus,DMN)、室 旁 核(paraventricular nucleus,PVN)、視交叉上核(suprachiasmatic nucleus,SCN)位于下丘腦內側區,其中ARC、VMN和DMN位于中部,PVN 和SCN 位于前區。外側區的外側下丘腦區(lateral hypothalamic area,LHA)神經核成為了下丘腦與其他部位聯絡的神經纖維出口[5],見圖1。
1.1 下丘腦弓狀核(ARC)在能量代謝的調節中心作用
控制能量穩態的中樞主要位于下丘腦及腦干,主要由下丘腦內的核團控制,如弓狀核(ARC)和室旁核(PVN)(見圖1)。來自血液循環中的激素分子,如瘦素、胰島素、GLP-1[6-7],以及營養物質[8],如脂肪酸可以通過下丘腦弓狀核等附近的不完整的BBB,影響這些特殊核團內神經元對能量攝入和儲存的信息感知,從而調節進食行為。此外,來自胃腸道迷走神經的傳入信息可以將食物消化、胃腸運動信息傳至腦干的孤束核,再由孤束核投射至下丘腦,因此我們可以感受到飽腹感或饑餓感,從而停止或增加進食[9]。然而,高級神經活動可以影響我們攝入行為,如通過獎勵機制促進或抑制。比如,人們普遍認為,可口的食物會導致我們對食物“成癮”而攝入更多,并且即使當一個人感到很飽或者能量滿足時,仍然控制不了進食,如果長期持續這種狀態,超越穩態飲食的享樂行為可能會導致體重增加及過度肥胖[10]。
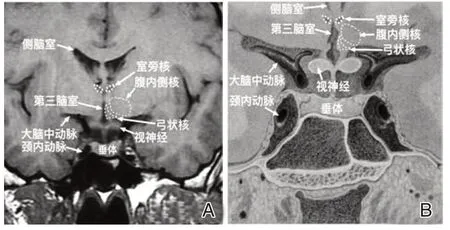
圖1 下丘腦神經核團-弓狀核、腹內側核和室旁核的解剖位置Figure 1 The anatomical location of the critical hypothalamic nuclei —the arcuate,ventromedial,and paraventricular nuclei
食物攝入可以由兩種相反的行為強烈驅動,厭食效應抑制進食和促食欲效應刺激食物攝入。解剖學和生理學證據表明下丘腦內的兩個主要不同的細胞系列[11]:表達神經肽Y(neuropeptide Y,NPY)和刺鼠相關肽(agouti-related peptide,AgRP)的促食欲神經元,表達前阿片-黑色素皮質素(proopiomelanocortin,POMC)和可卡因-苯丙胺調節轉錄物(cocaine-and-amphetamine regulated transcript,CART)的厭食神經元。下丘腦內POMC/CART 或NPY/AgRP 神經元的活動導致了厭食或促進食欲作用。POMC 神經元表達肽α-MSH,有導致厭食作用,通過對POMC的轉錄后處理,與PVN等處的二級神經元上的黑素皮質素受體3/4(melanocortin receptor-3/4,MCR3/MCR4)結合會影響新陳代謝,從而誘導厭食效應。一項研究表明,具有獲得性MC4R突變的MC4R基因敲除小鼠或人類個體與早發性嚴重肥胖相關。CART在ARC內與POMC共表達,抑制食物攝入,調節能量消耗。當CART 呈遞給二級神經元時,可以促進飽足感產生。注射CART 可減少嚙齒類動物的進食,使用CART 抗血清治療可減少吞咽不足。類似的,CART基因的突變或多態性與肥胖發病率的增加有關。我們之前的研究[12]表明,表達5羥色胺2C受體(serotonin receptor-2,5-HT2CR)的POMC神經元可被(serotonin,5-HT)激活,從而改善葡萄糖代謝。其活化可觸發從前體POMC 衍生的α-MSH 的產生增加。α-MSH作用于分布在脊髓中間外側核(intermediolateral nucleus,IML)內的MCR4,此處含有交感節前神經元,通過激動MCR4可以改善胰島素靶器官,如骨骼肌、肝臟、脂肪組織的胰島素敏感性。
1.2 下丘腦腹內側核(VMH)、杏仁核(amygdala)與血糖平衡有關
正常情況下,血糖受到升糖激素和胰島素調節,VMH 包含葡萄糖興奮神經元(glucose excited neuron,GE)與葡萄糖抑制神經元(glucose inhibited neuron,GI),低血糖使GI興奮,啟動反調節系統(counterregulatory response system),如下丘腦-垂體-腎上腺(HPA)軸、交感腎上腺髓質系統、胰高血糖素等,使肝糖原分解,促進糖異生,防止血糖進一步下降;而高血糖時GI抑制而GE 興奮,減少糖異生和升糖激素分泌,從而使血糖維持平衡,研究顯示這與葡萄糖激酶(glucose kinase,GK)、ATP 敏感的鉀離子通道(KATP)、AMP 激活地蛋白激酶(AMP activated protein kinase,AMPK)和一氧化氮(NO)等物質的參與有關[13-15]。我們發現低血糖反調節反應受到上一級神經中樞杏仁核的影響[16]。糖尿病低血糖癥定義為出現低血糖癥狀并且血糖低于3.9 mmol/L,而晚期糖尿病或1 型糖尿病病人可能因為長期低血糖環境,產生低血糖不敏感,甚至低于3.9 mmol/L也不產生低血糖反應,我們稱之為“低血糖相關自主神經調節障礙”(hypoglycemic associated autonomic failure,HAAF),研究顯示除了與胰島功能改變有關外,也與VMH 調節障礙有關,這些病人的糖尿病狀態俗稱“脆性糖尿病”(brittle diabetes)[17]。
1.3 前腦紋狀體的多巴胺釋放與獎賞系統及糖尿病外科代謝手術機制
獎賞機制途徑通過多巴胺(dopamine,DA)信號傳導發揮促進食物攝入的作用。有研究認為,病態肥胖現象可能是由于長期攝入過量可口食物導致多巴胺受體2(D2r)表達降低所致[18-19]。另一項研究發現,肥胖者的貪食癥可能反映了食物獎勵的“喜歡”(liking)和“想要”(wanting)之間平衡的改變[20]。TSAI等獨立發現的腹側被蓋區域(ventra tegmental area,VTA)是成癮中的重要核團,其解剖位置靠近紅核與黑質,并且存在較多的多巴胺能與5-HT 能神經元,主要成癮相關的通路包括從VTA 至前額葉皮質的中腦皮質通路(mesocortical pathway),以及從VTA 至腹側紋狀體中腦邊緣系統通路(meso-limbic pathway)[21]。VTA 的多巴胺及(γ-aminobutyric acid,GABA)神經通路與獎賞的調節有關[22-24]。最新的研究表明:嚙齒動物腹側紋狀體接受來自近端空腸營養神經信號的投射,背側紋狀體則接受十二指腸的營養神經投射,參與獎賞促進進食[25]。而十二指腸空腸旁路手術(duodenal-jejunal bypass,DJB)能消除十二指腸的糖誘導的背側紋狀體多巴胺釋放的影響,抑制葡萄糖的攝入,其中背側紋狀體含有表達多巴胺D1 受體的神經元,通過光遺傳學的方法可激活這些神經元,模擬糖誘導的多巴胺釋放,逆轉DJB 手術對進食的抑制;另外,耶魯大學的研究者們發現,胃腸的迷走神經傳入信號并非是左右對稱的,其中,背外側臂旁核(parabrachial nucleus)連接右側迷走性感覺性神經節至黑質,研究揭示一種通路由右側結狀神經節,臂旁核-黑質通路和背側紋狀體構成,是促膽囊收縮素(CCK)作用于食欲的必需的環節[26]。
1.4 腸道、肝臟、脂肪組織以及骨骼肌肉在能量代謝中的作用
人類的消化系統存在豐富的彌散神經內分泌系統,含有攝取胺前體脫羧細胞(APUD),多種與進食有關的內分泌激素產生于胃腸道,包括促進食欲的胃饑餓素(ghrelin),抑制食欲的胰高血糖素樣肽-1(GLP-1)、促膽囊收縮素(cholecystokinin,CCK)、酪酪肽(peptide YY,PYY)和葡萄糖依賴性促胰島素多肽(glucose dependent insulinotropic peptide,GIP)等;這些激素既可以通過局部迷走神經傳遞至孤束核(NTS),也可以通過血液循環到達下丘腦影響進食和體重[27]。消化道攝入食物后釋放的胃腸道信號,如GLP-1,對中樞神經系統能量和代謝的調節起著重要的負反饋作用,即消化道釋放的腸道激素構成了決定結束進食的重要因素。這些信號被稱為飽足信號,包括胃機械反應、腸道食物能量檢測和腸道肽激素釋放,如GLP-1、肽YY(PYY3-36)、膽囊收縮素(CCK)等。盡管操縱GLP-1等單個信號的水平可以對攝食、體重和血糖控制產生影響,但各種胃腸道、全身和中樞信號的綜合作用共同介導了食物攝入抑制的內源性控制。最近熱門研究腸道菌群(microbiota)也與能量調節及多種精神神經系統疾病、自身免疫性疾病發病相關[28],其內毒素(LPS)可以產生低度炎癥,肥胖與非肥胖人群腸道菌群報道有顯著差別,與代謝綜合征相關[29-30];但目前腸道菌群對代謝和疾病的貢獻大小、補充益生元對預防糖尿病的作用尚存爭議。
肝臟是人體代謝的中樞。大部分腸道激素以及胰島素都會受到肝臟的首過消除,外源的胰島素或GLP-1與機體內源性的激素最后的生理作用是否存在區別,這值得進一步的研究探索。非酒精性脂肪肝,現稱“代謝相關性脂肪肝病”(metabolic-associated fatty liver disease,MAFLD)也與胰島素抵抗、2 型糖尿病、痛風的發生密切相關[31-32]。我們發現肝臟產生的成纖維細胞生長因子21(fibroblast growth factor-21,FGF21)可以調節動物的體重及代謝[33]。
脂肪組織劃分包括棕色、米色以及白色脂肪組織,其中白色脂肪組織可以產生能量代謝有關因子,作用于下丘腦,如瘦素(leptin)、脂聯素(adiponectin)以及炎癥因子,目前一致認為脂肪組織炎癥(adipose tissue inflammation)與胰島素抵抗、高血壓發生相關[34-36]。兒童或幼年動物富含棕色脂肪組織,過去一直認為與產熱和減重有關,其受到中樞交感神經的支配,GLP-1受體激動劑可通過解偶聯蛋白(UCP-1)促進棕色脂肪組織產熱,達到體重減輕的作用[37]。米色脂肪組織雖然在正常基礎情況下含有少量的UCP-1,但在運動后骨骼肌產生的鳶尾素(irisin)或寒冷刺激后可以誘導其表達增加,促進白色脂肪組織分解及棕色化,因此在代謝中具有重要意義[38]。
近年來陸續有研究表明骨骼和肌肉也屬于內分泌器官,已發現多種骨骼產生的因子對糖代謝具有重要作用[39],有的甚至可以影響中樞神經系統MCR4[39],如骨骼中的骨鈣素(osteocalcin)具有促進胰島素分泌,改善胰島素抵抗作用,而我們在臨床研究中發現不管是1型還是2 型糖尿病人,尤其是在酮癥酸中毒情況下血骨鈣素水平偏低,提示骨骼活動與糖代謝有著密切聯系。骨骼肌可以產生多種肌肉因子(myokines),如irisin 和follistatins,尤其是在運動鍛煉后表達增加,可以顯著改善糖代謝[40]。
2 中樞神經系統對心血管自主神經系統的調節
心血管活動受到自主神經系統(autonomic nervous system)調節,該系統廣泛分布于下丘腦、腦干、中腦、杏仁核以及皮層等區域。研究發現參與高血壓及心衰的RAAS 系統組分可以表達于中樞神經系統,膠質細胞參與的炎癥反應與高血壓的病理生理過程相關[41]。心血管自主神經活動調節的相關核團包括:①參與高級活動控制的島葉(insular cortex)和內側前額葉(medial prefrontal cortex);②情緒狀態相關的杏仁核中央核團(central nucleus of amygdala)、終紋床核(bed nucleus of stria terminalis);③下丘腦啟動和協調自主神經活動、神經內分泌、行為反應和壓力反應,下丘腦在心血管活動調節意義已有大量研究證明[42],包括ARC及PVN,其中PVN 與心血管活動調節既包括至腦干的交感神經途徑,也包括至垂體前部與后部的神經內分泌途徑;④中腦導水管周圍灰質(periaqueductal gray,PAG),與自主神經、痛覺控制、壓力相關、侵略性、生殖相關食物行為有關;⑤腦橋臂旁Kolliker-Fuse核,傳遞內臟感覺信息至前腦,與呼吸、循環相關;⑥內臟感覺傳遞的孤束核(NTS);⑦延髓中間網狀區域尤其是腹外側延髓(VLM),包含自主神經的前運動神經元,調節心血管、呼吸運動。這些核團的活動構成機體即時的狀態,受到呼吸、睡眠覺醒、情緒狀態、注意力等影響[43]。
3 GLP-1RA和SGLT-2可以通過血腦屏障
血腦屏障(BBB)是維持中樞神經系統(CNS)正常生理狀態和代謝的動態屏障,但它也抑制某些藥物進入CNS 影響治療效果。在肥胖癥和2 型糖尿病病人中,BBB 密閉結構是動態變化甚至受損的[44-45],因此SGLT2i 和GLP-1RA 這兩種藥物有可能在這種情況下通過BBB,作用于下丘腦、腦干等相同的中樞神經系統通路,但是它們的控制模式可能不同[1,46]。目前的基礎和臨床研究表明,GLP-1RA能穿過BBB直接或間接作用于其受體,不僅可以降低血糖水平,還可以治療中樞神經系統疾病。利拉魯肽、艾塞肽和利昔那肽等GLP-1RA,已被證明通過被動擴散穿過BBB[47-49];司美格魯肽不能直接穿過正常的BBB,但可以與血清白蛋白結合,并通過腦室壁的室管膜Tanycyte細胞攝取[50]。目前雖然SGLT-2i類藥物通過BBB直接的動力學證據尚處于研究中,但已有間接研究表明其在患有神經疾病如癲癇、帕金森、Alzheimer、腦卒中的病人中具有神經保護作用。
4 GLP-1 受體激動劑(GLP-1RA)治療肥胖和2型糖尿病的神經內分泌靶點
人類的腸道L 細胞可自然分泌GLP-1[51],容易被DPP-IV 降解,只有很少部分通過血液循環到達BBB,可能通過局部的迷走神經傳入影響腦干的孤束核,釋放腦源性GLP-1,但目前對內源性GLP-1 是否屬于激素的本質尚存爭議[52]。KASTIN 等[53]觀察和分析了靜脈注射同位素標記的125I-[Ser8]GLP-1在ICR小鼠腦內流入率,發現標記的GLP-1 的快速流入腦實質屬于一種被動擴散。許多研究也發現中樞神經系統存在GLP-1 或是其代謝物的表達證據,尤其是腦干和嗅球存在GLP-1 的表達,而GLP-1 受體(GLP-1r)可以在腦內尤其是ARC 廣泛地分布[54],表明腸源性GLP-1 可能經被動轉運通過BBB影響攝食中樞,但作用可能不強。生理情況下,腸源性GLP-1 易被DPP-IV 降解,半衰期不足2 min,而藥理濃度的GLP-1不會完全被BBB血管內皮細胞上的DPP-IV 降解,從而可以大量進入腦實質,這也部分解釋了通過抑制DPP-IV 的藥物西格列汀、利格列汀等一類藥物對體重和食欲微弱影響的原因。但也有研究表明[55],GLP-1 只有在與GLP-1r 結合時才能進入大腦:GLP-1r介導的轉運分布在腦室周器(CVO)和下丘腦的其他部分,通過含窗孔毛細血管,和一種特殊的室管膜細胞伸長細胞(tanycyte)介導的轉運。因此GLP-1 靶向中樞核團可能存在多種機制,見圖2。
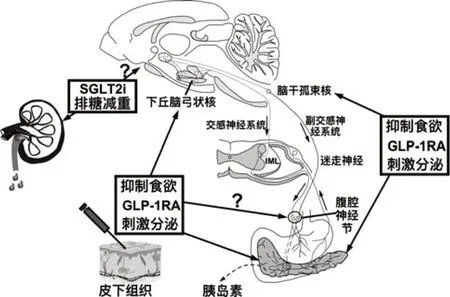
圖2 兩種降糖新藥的作用靶點比較Figure 2 Comparisons of the targets of two novel hypoglycemic medications
GLP-1進入中樞神經系統后可以作用于與能量穩態有關的多種核團[46,56-57],如ARC[55]、PVN[58]、NTS[59],影響進食、體重、產熱等能量平衡,而GLP-1 同時亦可影響獎賞中樞,如VTA、紋狀體、海馬等[60-62],繼而可以影響食物的動機、成癮、偏好等行為,調節能量的攝入。近幾年,科學家們圍繞GLP-1 如何影響能量代謝的核團進行了大量研究,主要是從消化系統迷走神經傳入、激活交感神經系統途徑,或GLP-1 通過下丘腦不完整的BBB 對下丘腦諸核團產生影響兩方面探討GLP-1 的機理及靶點。但目前被更多接受的觀點可能是GLP-1 通過多種途徑影響中樞神經系統[50]。GLP-1r 在ARC 激動導致的攝食和體重改變目前發現主要是通過影響POMC[55]與NPY[63]神經元的活動,而通過膜片鉗電生理實驗發現GLP-1RA 可以通過受體及下游帶有TRPC5混合離子通道的谷氨酸能神經元信號提高并興奮POMC 神經元,同時通過帶有ATP 敏感鉀離子通道(KATP)和TRPC5 的GABA 神經元抑制NPY神經元的興奮性從而促進POMC 的活動[64];也有研究提示激動支配POMC 的突觸前胞膜上的GLP-1r 可以促進POMC 的激活[65]。POMC 釋放的α-MSH 可以影響5-HT 攝食有關的信號通路調節能量攝入。有研究顯示T2D 病人下丘腦PVN 等區域的GLP-1r 表達明顯下降[66],這提示PVN 的GLP-1 信號減弱與T2D 的病理機制相關,PVN除了接受其它代謝途徑的信號[67],以及上級神經元的傳入信號外[58,68],激動PVN 的GLP-1r 可以通過增加CRH 神經元的依賴PKA 通路途徑信號,促進GluA1 AMPA 磷酸化及神經元胞膜的AMPA 受體表達,而PVN 的催產素、CRH 釋放以及交感神經等活動可以抑制進食[69]。NTS接收消化道迷走神經GLP-1等信號的傳入,已有實驗證明迷走神經阻滯或是選擇性迷走神經傳入神經損傷能減弱外周注射GLP-1 的進食效應[70],而通過c-Fos 表達研究發現某些代謝信號改變如長期高脂飲食,可能會損害迷走神經對GLP-1的敏感性[71]。此外,NTS 旁的室周器AP 具有不完整的BBB,高度血管化[72],同時也表達GLP-1r,而尾端孤束核表達GLP-1,卻不表達GLP-1r,我們推測腦部孤束核處的GLP-1 可能在外周注射GLP-1 激活迷走神經后介導了部分的減少進食作用,損毀這些室周器能減弱艾塞那肽誘導的孤束核c-Fos 表達,而迷走神經切除并不影響最后區的c-Fos 表達[73]。此外,通過組織學研究發現脊髓的IML 存在GLP-1r,并且可受NTS 的支配,而IML 可以影響靶代謝器官如肝臟、胰腺、骨骼肌等功能活動,是GLP-1 調節代謝器官的重要神經通路[12,74-75]。
5 SGLT-2i調節心血管交感神經活動的神經內分泌靶點
SGLT-2i最早來源于蘋果樹皮提取物根皮苷,其主要靶點是SGLT-1和SGLT-2,前者主要在小腸分布,以繼發性主動轉運方式吸收葡萄糖,而腎臟近曲小管大量分布的SGLT-2主要負責原尿中90%葡萄糖重吸收,但根皮苷選擇性低,不良反應較多;目前改良的SGLT-2i 主要作用于SGLT-2,和少量的SGLT-1[76],由于不依賴于胰島素,因此可以有效的排糖和促進體重減輕。SGLT-2i 在眾多的臨床試驗中被證明能顯著改善慢性心衰和減少心血管3P-MACE事件[77],除了滲透性利尿作用,還可能與有效改善心衰的神經內分泌狀態有關,如抑制交感神經過度激活,抑制RAAS激活[78]。腎臟近曲小管的SGLT-2 表達量與機體的交感活性強度成正比,在多數肥胖和2型糖尿病病人或動物模型中,可以發現交感過度激活狀態,以及SGLT-2在腎近曲小管過表達[79],這說明肥胖和2型糖尿病病人體內存在交感過度激活狀態,而SGLT-2i 可以減少這種狀態并防止并發癥出現。目前對SGLT-2i作用于中樞交感神經系統的研究不多,但也有不少數據支持這種猜測,如SGLT-2i 可以改善藥物所致癲癇發作[80],改善中樞下丘腦等處的神經炎癥,減輕代謝綜合征表現。SA-NGUANMOO等[81]同樣發現達格列凈能通過改善中樞線粒體氧化應激及胰島素信號,影響炎癥及凋亡,并且可以改善認知功能衰退,如恢復海馬的突觸可塑性,炎癥信號、胰島素信號的改善是海馬功能改善的主要原因,促進認知功能恢復的同時也可能通過HPA軸降低血皮質醇的濃度,一定程度上影響代謝綜合征導致的認知功能受損及血壓波動[82]。也有研究提示當將SGLT-2i 輸入老鼠的腦部后,可以明顯促進進食代償能量不足[83],也與產熱神經通路的調節改變有關[84]。SGLT-2i 對心血管活動的調節具有明顯的中樞效應,如有研究發現,使用魯格列凈可以改善SHRcp 大鼠的血壓節律[85],可能機制是影響下丘腦視上核活動;而現有間接研究提示SGLT-2i 可以改善諸如Alzheimer、帕金森等退行性疾病,病理機制包括影響Alzheimer膽堿酯酶活性[86],通過對接研究發現SGLT-2i也可能影響帕金森病A2A腺苷受體[87];在這類疾病中,腦部的BBB可能因為氧化應激等活動出現結構改變,因此SGLT-2i 在這種狀態下有極大可能性通過受損的BBB[88]。此外研究也提示SGLT-2i 可以影響腸道菌群的改變,影響腦腸軸GLP-1、Ghrelin 等激素分泌,改善代謝綜合征和心血管系統獲益[89]。
我們之前的研究發現中樞SGLT-2 可以分布于下丘腦、中腦、杏仁核、腦干等與自主神經活動相關的區域,SGLT-2i達格列凈可以改變小鼠的血壓、心率,激活腦部c-Fos 表達[90]。SGLT-2i 具有分子小、脂溶性特點,可能通過BBB。而SGLT-2i 改善心衰的神經內分泌機制仍不甚清楚,有待進一步研究。
6 小結
本文主要總結了2型糖尿病發病的神經內分泌機制,而兩種降糖新藥可以改善相關的神經內分泌狀態,與傳統的降糖藥物不同,GLP-1RA 主要影響肥胖的中樞代謝有關的靶點,而SGLT-2i 則主要針對心血管交感神經活動。通過介紹能量代謝與心血管活動調節,以及兩種降糖藥物的神經內分泌靶點,希望有助于更多靶向藥物的研發。
(利益沖突:無)
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
