基于木香藥材-標準湯劑-配方顆粒的指紋圖譜及含量測定研究
2022-07-27 02:58:36韓慧琴狄慧荊燕燕楊桂英尹貽珍
世界最新醫學信息文摘 2022年24期
韓慧琴,狄慧,荊燕燕,楊桂英,尹貽珍
(張家口市食品藥品檢驗中心,河北 張家口 075000)
0 引言
木香(Aucklandiae Radix )來源于菊科植物木香的干燥根。木香可以行氣止痛、調中導滯,如果患者有脾胃氣滯,脘腹脹滿或者出現疼痛,并且伴有噯氣、惡心、嘔吐的患者,可以應用木香進行治療[1]。木香在臨床上是比較常用的一種中藥材,古代醫家將其列為上品[2]。其主要含有萜類,蒽醌類,生物堿類,黃酮類等。目前關于木香的研究多為含量測定[3]、藥理作用[4]和藥材的指紋圖譜[5]等方面。關于木香配方顆粒的報道較少。
中藥配方顆粒是通過現代化的制藥手段,以中藥飲片為初始原料,用水煎煮提取、放冷過濾、減壓干燥、制粒成形,其藥效物質、性味歸經、功能主治和傳統中藥湯劑一致,既能保證中醫辨證論治的特點、又可以靈活加減,方便患者服用,衛生有效[6-8]。國家藥監局發布2021年第22 號公告,結束中藥配方顆粒試點工作,說明配方顆粒具備很多優點,并且已被接受,故國家鼓勵生產配方顆粒。但是從質量標準來看,目前檢驗項目還不是很全面,特別是安全性有效性方面還有待提高。因此,課題組參考有關文獻[9,10]采用HPLC 法建立了指紋圖譜及含量測定方法,可為木香配方顆粒的質量控制提供參考依據,可為其真偽優劣鑒別提供參考手段。
1 儀器與試藥
LC-30AT 型高效液相色譜測定儀(配備二極管陣列檢測器)(日本島津公司); e2695-2998(配備二極管陣列檢測器)型高效液相色譜儀(美國Waters 公司);國家藥典會“中藥指紋圖譜相似度評價軟件(版本號為2012.130723)”;統計軟件(SPSS 19.0);BT125D 型精密十萬分之一電子天平(德國sartorius,分度:0.01mg);DTC-27 型靜音型超聲波清洗機(湖北鼎泰高科有限公司);木香烴內酯對照品(批號111524-201911,99.9%)和去氫木香內酯對照品(批號111525-201912,99.5%)均來源于中檢院;15 批飲片分別購自四川(5 批)、云南(5 批)、廣東(3 批)和廣西(2 批)5 個省區,自制標準湯劑,批號分別為TJ01-TJ15。10 批樣品由2 個生產廠家提供,批號見表3。
色譜純乙腈和色譜純磷酸;超純水。
2 方法與結果
2.1 色譜條件
色譜柱信息:Scienhome YWG C18( P/N:96182540);Welch,XB-CN(Part Number:00205-31043);InertSustain AQ-C18(P/N:5020-89731);GL Sciences,Inertsil ODS-3(Cat.No.5020-01732);Agilent EXTEND C18(P.N.5188-5292),5 個廠家的色譜柱均為粒徑5μm,長250mm,內徑4.6mm。流動相:乙腈(A)-0.05%磷酸溶液(B)梯度洗脫(0~5min,32%~50%A;5~35min,50%~68%A;35~36min,68%~32%A,36~46min,32%A;在225nm 的波長處檢測;30 ℃的柱溫;體積流量:1.0mL/min,進樣體積10μL;儀器顯示的去氫木香內酯的理論塔板數應不低于3000。
2.2 標準湯劑的制備
取15 批次木香藥材各200g,加水2000mL 煎煮1 小時,過濾,取濾渣加水1400mL 煎煮1 小時,過濾,合并濾液,低溫蒸干,15 批次平均出膏86.3g,平均出膏率為43.2%。
2.3 對照品溶液
稱取去氫木香內酯對照品、木香烴內酯對照品適量,精密稱定,分別加甲醇溶解稀釋成每1mL 含去氫木香內酯194.4μg、木香烴內酯213.5μg 的溶液,用孔徑為0.45μm 的nylon 過濾膜過濾即得。
2.4 供試品溶液
分別精密稱取木香對照藥材0.25g;標準湯劑和配方顆粒,研細(四號篩)約0.5g;置150mL 帶有瓶塞的錐形瓶中,用大肚吸管精密量取甲醇50mL,加入錐形瓶中,用電子天平稱定重量后,超聲波提取( 功率260 瓦,頻率5 萬赫茲)30min,放冷,用0.45μm 的nylon 過濾膜過濾,取續濾液,即得。
2.5 含量測定方法學考察
2.5.1 專屬性
取“2.3~2.4”項下的溶液各10μL 注入液相色譜儀,結果去氫木香內酯峰與其它峰的分離度均大于1.5,且峰型好,表明專屬性強,見圖1。

圖1 HPLC 色譜圖(A)去氫木香內酯對照品(B)木香烴內酯和去氫木香內酯混合對照品(C)木香藥材(D)木香配方顆粒(E)標準湯劑
2.5.2 線性關系考察
精密量取“2.3”項下去氫木香內酯對照品溶液,用移液槍分別精密量取4、10、50、80、200、400μL 置1mL 的量瓶中,分別加甲醇稀釋至刻度,配制成系列濃度的標準溶液。分別進樣10μL,以對照品的進樣濃度(μg/mL)A 為橫坐標,以去氫木香內酯峰面積積分值B 為縱坐標進行線性回歸,建立標準曲線,去氫木香內酯的回歸方程為A=15450 B-1151.4 (相關系數為0.9998) ,線性范圍為0.656~65.6μg/mL。
2.5.3 精密度
取“2.4”項下的同一供試品溶液(批號:20090511),連續進樣6 次,進行測定。去氫木香內酯的RSD 為0.8%,小于2.0%,表明儀器的精密度良好。
2.5.4 重復性
取同一批木香配方顆粒(批號:20090511),研細,照“2.4”項下的方法同時稱取6 份供試品,用同一方法配制成供試品溶液,進樣分析,計算去氫木香內酯的含有量RSD 為1.2%,小于2.0%,結果顯示該提取方法和儀器方法的重復性良好。
2.5.5 準確度
分別稱取已知含量的木香配方顆粒(批號:20090511)研細,混勻,取9 份,每份約0.25g,精密稱定,每三份為一組,分別按樣品中含量的50%,100%,150%的量加入去氫木香烴內酯對照品,按“2.4”項下的方法進行提取制備。按上述“2.1”項下描述的色譜條件測定并計算回收率。結果回收率 RSD 值為0.7%,小于3.0%,見表1。表明該提取方法可行。
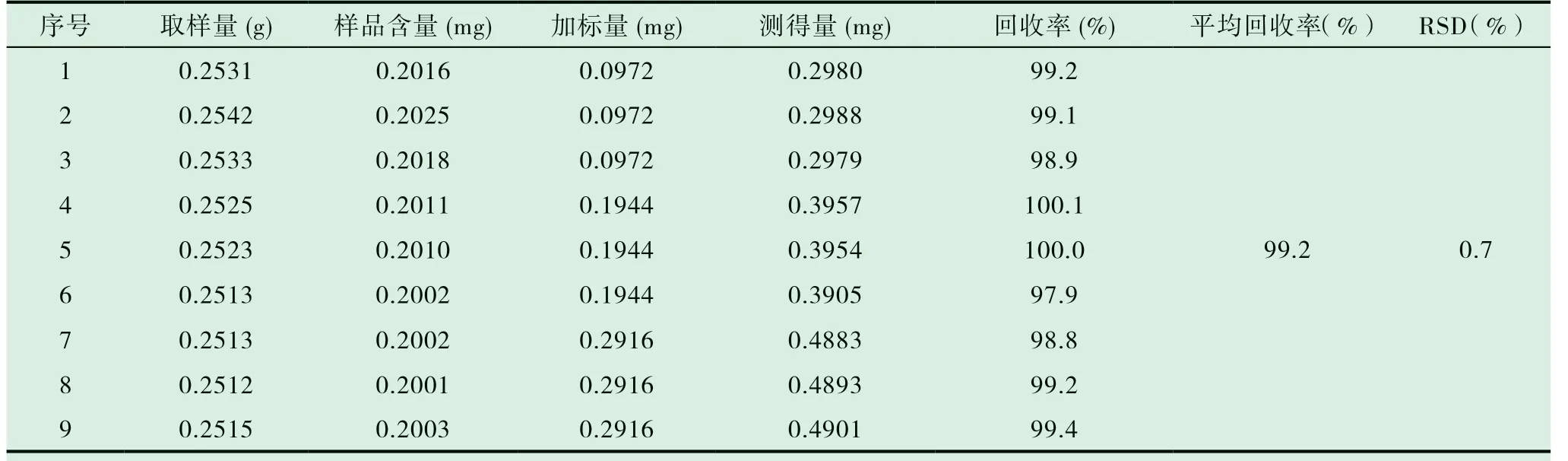
表1 回收率實驗結果(n=9)
2.5.6 穩定性
取同一批木香配方顆粒(批號:20090511)的供試品溶液,在室溫(26℃)下放置,分別于0、1、2、3、6、12、18、24、48h 按上述“2.1”項下描述的色譜條件進樣9 次,結果去氫木香內酯含量的RSD 為0.8%,低于3.0%,表明制備好的溶液在26℃下可以放置48h。
2.5.7 耐用性
分別采用島津LC-30AT 型和Waters e2695-2998 型高效液相色譜儀,采用Scienhome YWG、Welch XB-CN、InertSustain AQ-C18、GL Sciences和Agilent EXTEND 5 個廠家的色譜柱按擬定的方法測定,結果去氫木香內酯含量的RSD 為1.8%,表明不同廠家的儀器和色譜柱檢測效果良好。
2.5.8 樣品的測定
取10 批木香配方顆粒,按前述“2.4”項下描述的方法制備供試品溶液,按前述“2.1”項下描述的色譜條件測定并計算樣品中去氫木香內酯的含量,結果見表2。

表2 樣品測定結果(n=2)
2.6 指紋圖譜方法學考察
2.6.1 精密度
取同一份批號為20090511 的木香配方顆粒供試品溶液,按前述“2.1”項下描述的色譜條件批處理進樣6 次,進行測定,以3 號色譜峰(去氫木香內酯)為參照計算6 次色譜圖中每個共同出現的色譜峰的相對保留時間、相對峰面積值的RSD,結果分別為0.7%,1.1%。表明7 個共有峰的相對保留時間RSD 與相對峰面積RSD 均小于2.0%,說明精密度良好。見圖3。
2.6.2 重復性
取同一批木香配方顆粒(批號:20090511),混勻,0.5g 共6 份,精密稱定,照“2.4”項下的提取方法配制供試品溶液,按前述“2.1”項下描述的色譜條件批處理進樣分析,計算相對峰面積值和相對保留時間的RSD 值。結果7 個共有峰與參照峰的相對峰面積值和相對保留時間的RSD 均低于2.0%,表明方法重復性良好。
2.6.3 穩定性
取同一份木香配方顆粒供試品溶液(批號:20090511),在室溫(26℃)下放置,分別于0、1、2、3、6、12、18、24、48h 按上述“2.1”項下描述的色譜條件進樣9 次,以去氫木香內酯峰為參照峰,計算7 個共有峰的相對保留時間和相對峰面積的RSD,結果均小于2.0%,表明制備好的溶液在26℃下可以放置48h。
2.6.4 耐用性
分別采用LC-30AT 型和e2695-2998 型高效 液 相 色 譜 儀,采 用Scienhome YWG、Welch、InertSustain、GL Sciences、Agilent EXTEND 5 個牌子的色譜柱按擬定的方法測定,以去氫木香內酯峰為參照峰,計算7 個共有色譜峰的相對峰面積值和相對保留時間的RSD,結果均低于2.0%,結果顯示方法的儀器及色譜柱耐用性良好。
2.7 指紋圖譜的建立
2.7.1 分別精密吸取不同批次(S1-S10)的木香配方顆粒供試品溶液、混合對照品溶液、標準湯劑溶液和對照藥材溶液批處理進樣10μL,按“2.1”的色譜條件測定,記錄色譜圖,見圖1。將10 批樣品的實驗數據導入國家藥典委員會的中藥色譜指紋圖譜相似度評價系統(2012),采用中位數法對10 批樣品的指紋圖譜進行峰點校正,數據匹配分析,生成共有模式圖及對照指紋圖譜,見圖2、3,確認7 個共有峰為特征峰,并用對照品圖譜進行定位,確定2 號峰為木香烴內酯,3 號峰為去氫木香內酯,見圖3。以3 號峰為參照峰,計算各共有峰的相對保留時間和相對峰面積,結果見表3、4。10 批木香配方顆粒與對照指紋圖譜的相似度分別為0.989,0.989,0.982,0.990,0.989,0.989,0.972,0.972,0.973,0.980 相似度評價表明,10 批供試品指紋圖譜和對照品指紋圖譜的相關性良好,且7 個共有峰與標準湯劑中的峰都能一一對應。對比相似度較小樣品與其他樣品的指紋圖譜,發現其共有峰相對保留時間比較一致,主要差異表現在色譜峰的面積上,即含量的差異,為不同廠家的樣品。表明廠家不同樣品的質量存在一定的差異。
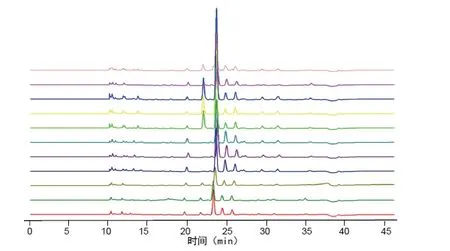
圖2 10 批樣品指紋疊加圖譜
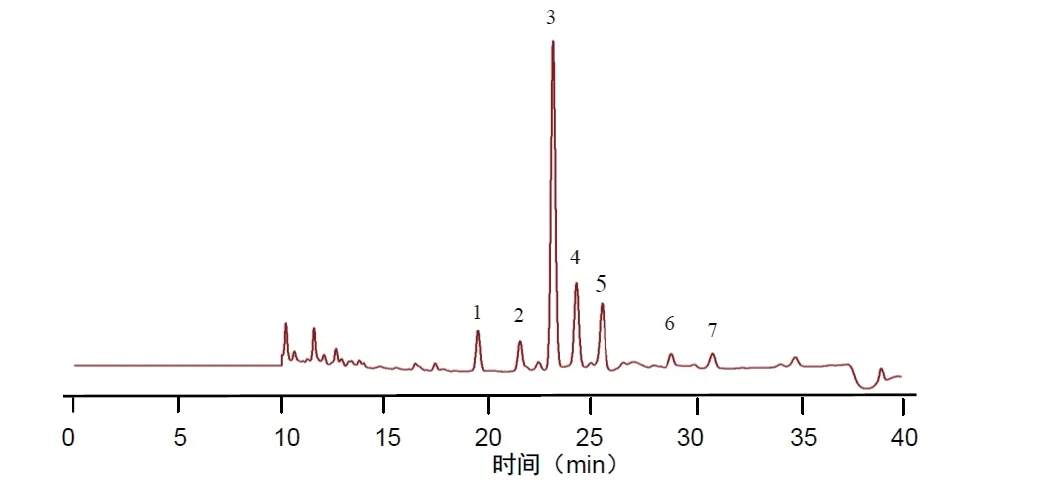
圖3 對照指紋圖譜
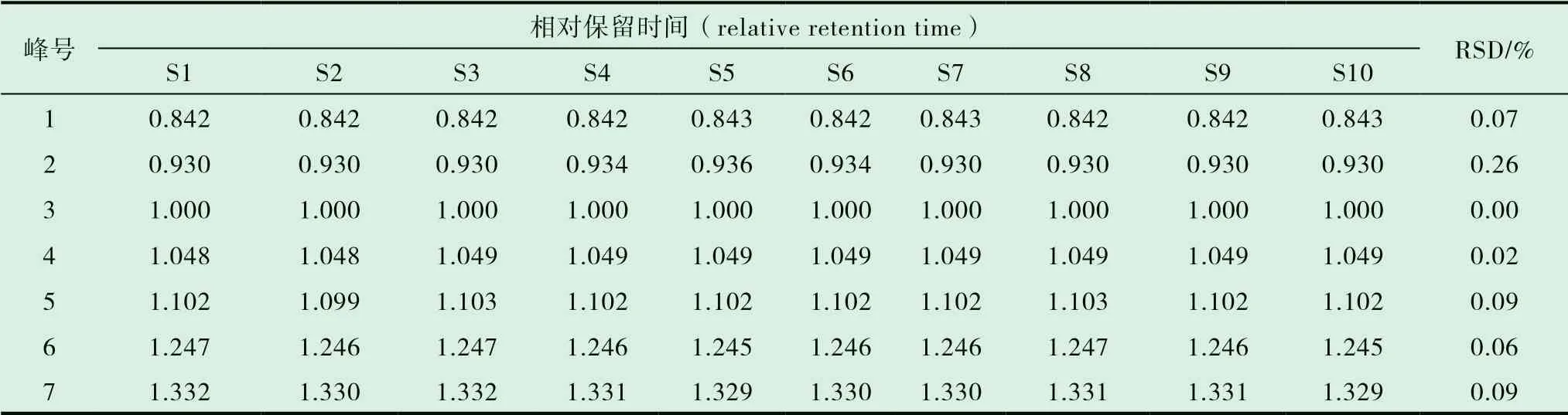
表3 10 批木香配方顆粒共有峰的相對保留時間

表4 10 批木香配方顆粒共有峰的相對峰面積
2.8 化學模式識別
2.8.1 聚類分析
以指紋圖譜中標定的7 個共有峰的相對峰面積為變量,采用SPSS 19.0 軟件中的平方Eudidean距離對10 批樣品進行系統聚類分析,結果見圖4。根據聚類分析結果,以類間距為指標,當聚類標定距離定為25 時,10 批樣品聚為一大類,當聚類重新標定距離為4 時,樣品S7、S8、S9 聚為第二小類,其余樣品聚為第三小類。該分析與相似度評價結果一致。
2.8.2 主成分分析
對10 批樣品進行主成分分析,以指紋圖譜中標定的7 個共有峰峰面積為分析變量,將其導入SPSS 19.0 統計軟件進行主成分分析,得到主成分特征值和方差貢獻率,見表5,圖5。可見前2 個主成分的特征值均大于1,累積貢獻率為98.453%,基本可以反映各主成分的全部信息。
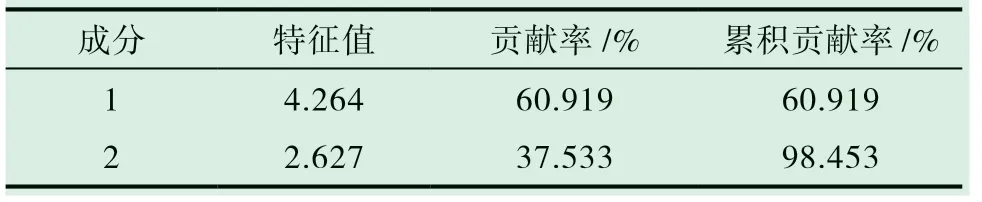
表5 主成分特征值和貢獻率
2.8.3 綜合評價
以各主成分對應貢獻率為權重系數,計算10 批樣品的主成分得分和綜合得分,并進行排序,結果見表6。綜合得分排名為S9>S5>S6>S4>S8>S7>S1>S2>S10>S3。其中生產日期較近的批次主成分得分較高,生產日期較遠的批次相對較低,另外,不同樣品批號中的藥材產地不同也導致其質量有所不同。
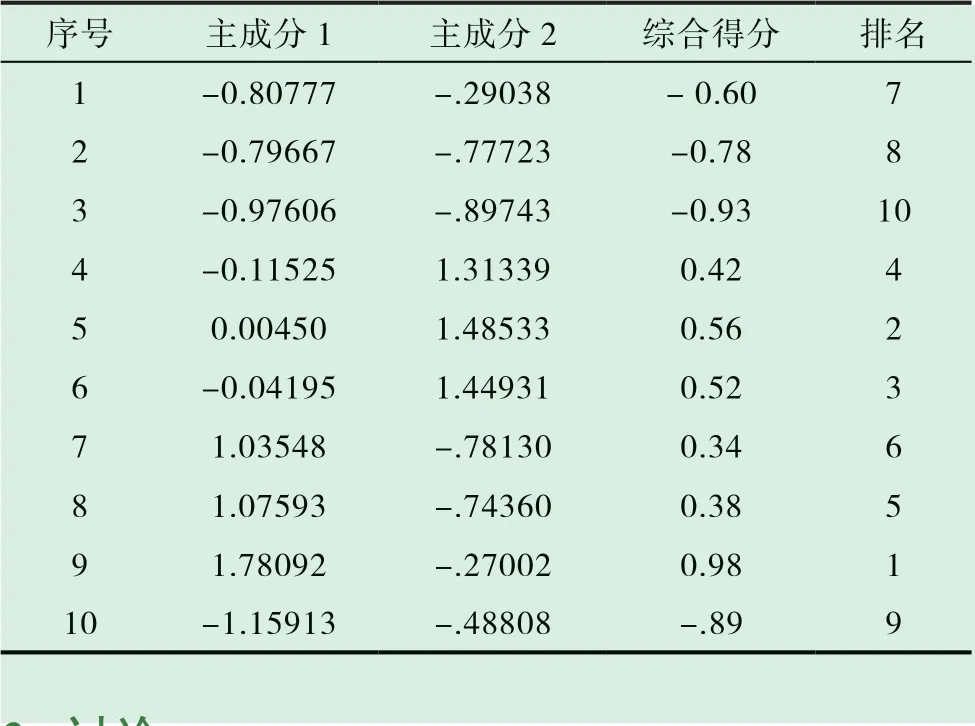
表6 10 批樣品的主成分得分、綜合得分和排名統計
3 討論
實驗過程中比較了甲醇、乙醇、70%甲醇、70%乙醇作為提取溶劑,結果甲醇提取效率最好;考察了甲醇-水、乙腈-水、乙腈-0.05%磷酸等流動相體系,考察了高效液相色譜儀和超高效液相色譜儀的檢測效果,最終確定了文中的方法,該條件下色譜峰信息豐富,分離度好,穩定性高,可以作為控制木香配方顆粒的方法。
本實驗通過高效液相色譜法對比研究木香藥材,標準湯劑和市售配方顆粒,找到指標成分進行含量測定,并進行指紋圖譜的研究及相似度評價,對木香配方顆粒進行了定性、定量評價,相似度都大于0.97,表明其質量比較一致,且與標準湯劑圖譜也一致,可見該方法更具可靠性。但是通過與藥材圖譜進行對比后發現制成配方顆粒后木香烴內酯的含量下降較多,故沒有選取木香烴內酯進行含量測定。而且其他峰也有變化,表明藥材與配方顆粒在成分方面有一定的差異,還有待研究。
采用聚類分析將10 批樣品分為3 類,主成分分析采用降維模式,將反映HPLC 指紋圖譜的多維特征參數用2 個主成分表示,可為木香配方顆粒質量評價提供參考。
