3種常見中藥材中綠原酸類成分提取的“逆溶解度”現象△
2022-07-06 01:47:40陳樂郭璐娟康利平朱蕓蕓陳昌婕于金倩杜鴻志劉大會
中國現代中藥 2022年6期
陳樂,郭璐娟,康利平,朱蕓蕓,陳昌婕,于金倩,杜鴻志,*,劉大會*
1.湖北中醫藥大學 中藥資源中心,湖北 武漢 430065;2.中國中醫科學院 中藥資源中心 道地藥材國家重點實驗室培育基地,北京 100700;3.齊魯工業大學(山東省科學院)山東省分析測試中心,山東 濟南 250014
中藥化學成分復雜多樣,其成分的復雜性構成了功效的多樣性[1]。根據中藥中各種化學成分在溶劑中的溶解性質選擇合適的提取溶劑,可顯著提高中藥中有效成分的提取率。中藥提取主要遵循“相似相溶”原理,即選擇和溶質極性相近的溶劑進行提取[2],這對中藥配方顆粒的制備、植物化學成分的分離等具有重要意義。綠原酸類成分是咖啡酸與天然奎寧酸的結合物,存在于多種高等植物中[3]。近年來,人們對綠原酸類化合物的關注日益增加,原因除了其具有酚酸、黃酮等酚類化合物常見的生物活性(抗菌、抗病毒、抗氧化、抗炎、抗腫瘤作用)及相對較低的不良反應外[4-6],綠原酸類化合物還被證實能降低血糖、治療輕度高血壓和降低膽固醇水平等[7-8]。因此,隨著關于綠原酸類成分研究的逐年增多,有必要對中藥中綠原酸類成分在不同溶劑中的溶解性、提取過程中含量變化規律等展開研究,以探尋中藥中各綠原酸類成分的最佳提取條件。
菊科蒿屬植物艾葉Artemisia argyiLevl.et Vant.、菊科菊屬植物菊花Chrysanthemum morifoliumRamat.及忍冬科忍冬屬植物金銀花Lonicera japonicaThunb.均是中國傳統大宗常用中藥材。近年來,在醫療保健、養生美容及動物飼料等相關領域備受青睞,是目前綠原酸類化合物的重要來源[3,9-10]。本課題組在前期實驗中發現,艾葉提取過程中存在“逆溶解度”現象(即中藥材中綠原酸類化合物在某種溶劑中的提取率與其在該溶劑中的溶解性能相反的現象)。例如,艾葉中綠原酸類成分咖啡酸溶解性質為微溶于冷水,易溶于乙醇,而艾葉在超聲提取時咖啡酸在純水中的提取率卻顯著高于乙醇,與自身溶解特性相反。在此基礎上,本研究系統測定了艾葉、菊花和金銀花3 種中藥材中主要綠原酸類成分在純水、乙醇的溶解度,并利用超聲輔助提取法和加熱回流提取法比較了3 種中藥材中綠原酸類成分在水和乙醇中提取率的差異,揭示了中藥在不同科屬、不同提取條件下的“逆溶解度”現象,同時探究了各綠原酸類成分在不同體積分數乙醇中的提取規律,以期為綠原酸類成分的提取分離、中藥配方顆粒制備工藝的優化及深入挖掘應用價值提供參考。
1 材料
1.1 儀器
1260 型超高效液相色譜儀(美國Agilent 公司);PS-40 型超聲波清洗器(深圳超藝達科技有限公司);ZNHW 型智能恒溫電熱套(天津工興實驗室儀器有限公司);EX125DZH 型十萬分之一電子天平(上海奧豪斯儀器有限公司);WP-UPGX 型超純水機(上海研承儀器有限公司)。
1.2 試藥
對照品新綠原酸(5-CQA,批號:DSTDX001501)、綠原酸(3-CQA,批號:DST200822-021)、咖啡酸(CA,批號:DST191030-013)、隱綠原酸(4-CQA,批號:DSTDY003501)購于德斯特生物科技有限公司;對 照 品 異 綠 原 酸B(3,4-diCQA,批 號:Y06911812012)、異綠原酸A(3,5-diCQA,批號:Y06801911011)和異綠原酸C(4,5-diCQA,批號:Y07011805016)購于成都瑞芬思生物科技有限公司,所有對照品純度均>98%;乙腈為色譜純(德國默克公司);無水乙醇、甲酸均為分析純(國藥集團化學試劑有限公司)。
2 份金銀花樣品、4 份艾葉樣品和4 份菊花樣品均于2020 年采摘,保存于湖北中醫藥大學中藥資源中心,由湖北中醫藥大學劉大會教授鑒定均為正品。樣品信息見表1。
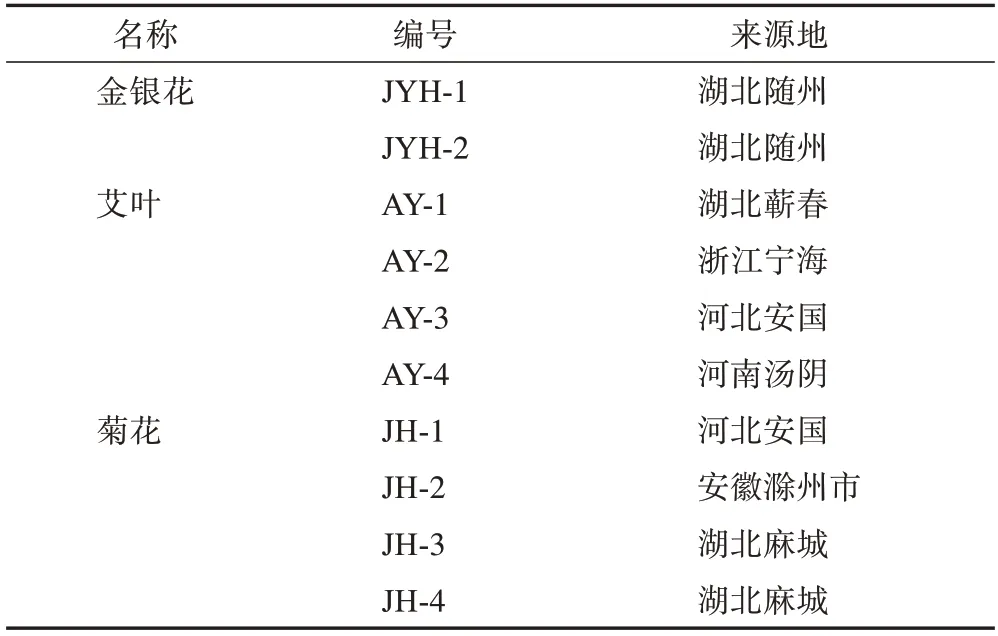
表1 金銀花、艾葉、菊花樣品信息
2 方法
2.1 超高效液相色譜法(UHPLC)條件
參照文獻[11]的UHPLC 色譜條件對艾葉、菊花和金銀花樣品中主要綠原酸類成分的含量進行測定,以0.1%甲酸水(A)-0.1%甲酸乙腈(B)進行梯度洗脫(0~0.3 min,2%~5%B;0.3~1.0 min,5%B;1.0~7.0 min,5%~20%B;7.0~9.0 min,20%B;9.0~9.5 min,20%~25%B;9.5~12.5 min,25%~28%B;12.5~18.0 min,28%~40%B;18.0~18.3 min,40%~80%B;18.3~21.0 min,80%~98%B;21.0~24.0 min,98%B);流速為0.5 mL·min-1;柱溫為40 ℃;進樣量為1 μL;檢測波長為330 nm。對該法進行儀器精密度、方法重復性、穩定性及加樣回收率方法學考察。
2.2 對照品溶液的制備
精密稱取7個對照品適量,用甲醇溶解、定容,制得5-CQA、3-CQA、CA、4-CQA、3,4-diCQA、3,5-diCQA、4,5-diCQA 質量濃度分別為7.37、12.71、29.11、3.73、315.56、545.00、358.33 mg·L-1的混合對照品溶液。
2.3 綠原酸類成分的溶解度測定
稱取各對照品適量,分別以純水和乙醇為溶劑,置室溫下充分振搖后超聲60 min,使其達到溶解平衡狀態,稀釋一定倍數后按2.1項下條件進行測定,分別測定7個綠原酸類成分在2種溶劑中的溶解濃度。
2.4 不同提取方法下綠原酸類成分在2 種溶劑中提取率的測定
2.4.1 超聲輔助提取法 將3 種中藥材粉碎,過一號篩,精密稱取各樣品粉末0.1 g 于20 mL 量瓶中,分別加入純水、乙醇定容至刻度線,稱定質量,放置過夜,超聲60 min,冷卻至室溫后用相應溶劑補足減失質量,靜置,經0.22 μm 微孔濾膜濾過后按2.1項下條件進行測定。
2.4.2 加熱回流提取法 精密稱取各樣品粉末5.0 g 于2000 mL 三頸燒瓶中,分別加入純水、乙醇1000 mL,稱定質量,放置過夜,置電熱套中回流提取60 min,冷卻至室溫后用相應溶劑補足減失質量,靜置,經0.22 μm 微孔濾膜濾過后按2.1 項下條件進行測定。
2.5 綠原酸類成分在不同體積分數乙醇中的提取規律
精密稱取各樣品粉末0.1 g 于20 mL 量瓶中,分別加入不同體積分數乙醇(0、25%、50%、75%、100%)定容至刻度,稱定質量,放置過夜,超聲60 min,冷卻至室溫后用相應體積分數的乙醇補足減失質量,靜置,經0.22 μm微孔濾膜濾過后按2.1項下條件進行測定。各對照品及樣品色譜圖見圖1。
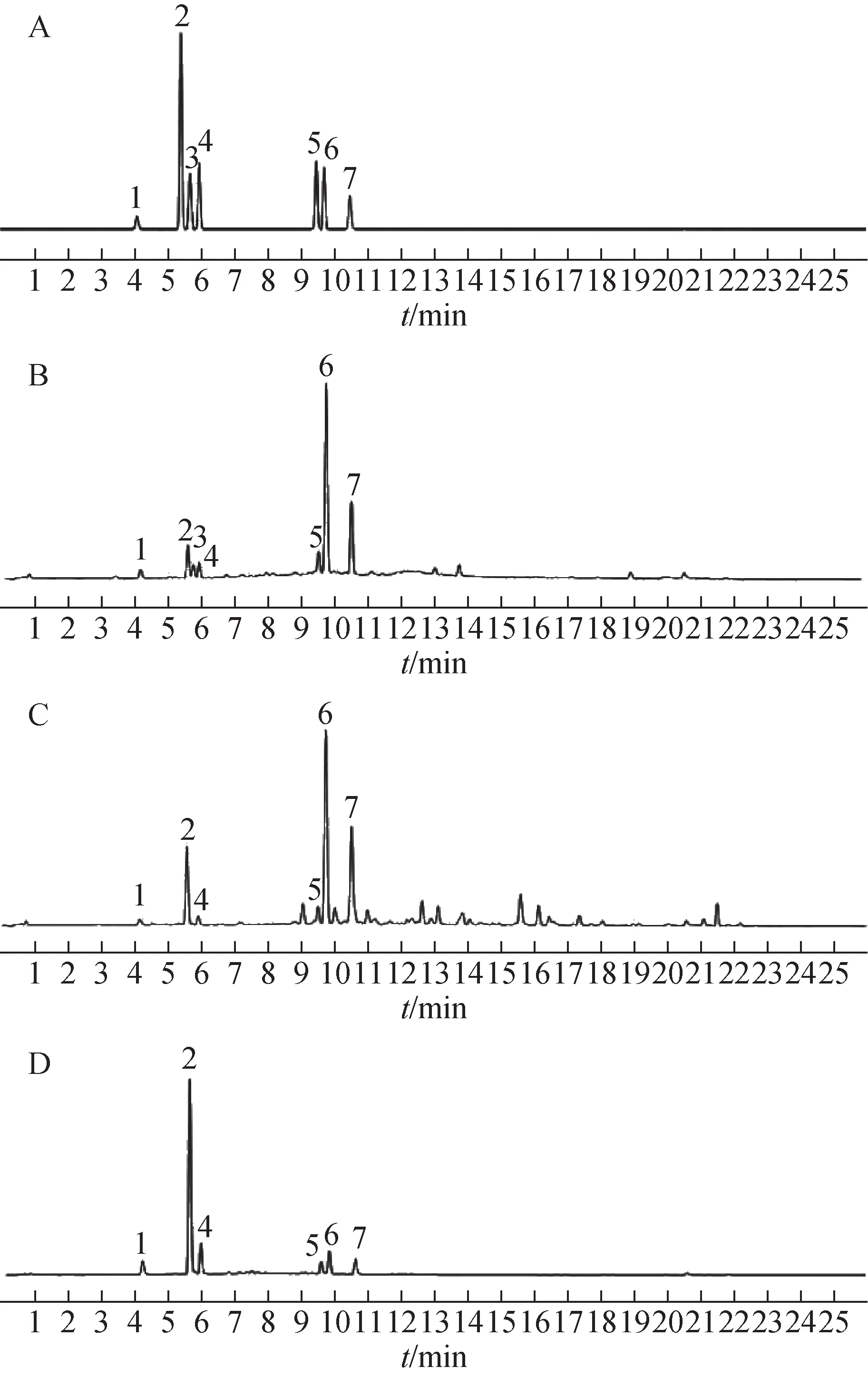
圖1 混合對照品及樣品在水溶液中超聲提取時的UHPLC圖
2.6 方法學考察
2.6.1 線性關系考察 精密吸取2.2 項下混合對照品溶液適量,稀釋成系列質量濃度的混合對照品溶液,分別按2.1 項下條件測定,以各對照品的質量濃度為橫坐標(X)、相應的峰面積為縱坐標(Y)進行線性回歸分析,見表2。
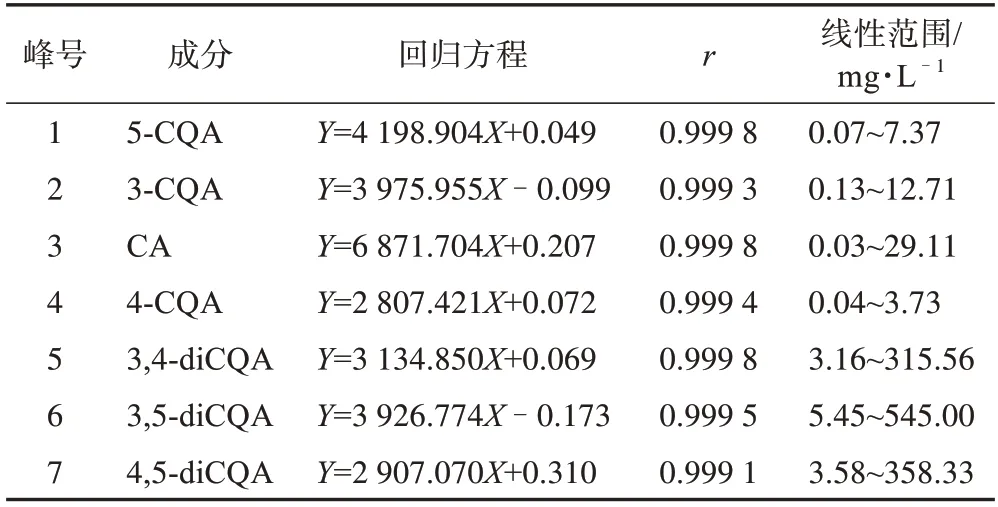
表2 7個綠原酸類成分的線性關系
2.6.2 精密度試驗 分別取同一份混合對照品溶液,按2.1 項下色譜條件連續進樣6 次,結果5-CQA、3-CQA、CA、4-CQA、3,4-diCQA、3,5-diCQA、4,5-diCQA峰面積的RSD分別為1.20%、1.27%、0.83%、1.15%、0.92%、1.05%、1.07%,說明儀器精密度良好。
2.6.3 穩定性試驗 稱取同一批粉末樣品,分別按2.4.1 和2.4.2 項下方法制備的樣品,于0、2、4、8、12、16、24、48 h進樣,按2.1項下色譜條件進樣分析,計算樣品中7 個對照品色譜峰峰面積的RSD,超聲輔助提取時峰面積的RSD 分別為1.2%、1.1%、1.3%、1.2%、1.2%、1.1%、1.4%,加熱回流提取時峰面積的RSD 分別為1.3%、1.2%、1.4%、1.2%、1.5%、1.3%、1.4%,說明樣品在48 h內穩定性良好。
2.6.4 重復性試驗 稱取同一批粉末樣品0.1 g,平行6 份,分別按2.4.1 和2.4.2 項下方法制備樣品后按2.1項下色譜條件進樣分析,計算樣品中7個綠原酸類成分含量的RSD,超聲輔助提取時含量的RSD 分別為1.4%、2.1%、1.7%、1.5%、1.5%、1.2%、1.3%,加熱回流提取時含量的RSD 分別為1.6%、1.4%、1.2%、1.5%、1.2%、1.4%、1.2%。
2.6.5 加樣回收率試驗 精密稱定已知含量的同一批粉末樣品0.1 g,平行9份,分別按1.0∶0.8、1∶1、1.0∶1.2 依次精密加入單一對照品溶液,每個比例3 份,然后按2.4.1 和2.4.2 項下方法制備樣品后按2.1項下色譜條件進樣分析,計算各對照品的加樣回收率。7個對照品溶液的在超聲輔助提取時加樣回收率分別為96.9%、97.3%、102.5%、100.4%、98.6%、99.2%、101.3%,對應的RSD 分別為1.6%、2.2%、1.8%、0.9%、2.1%、1.1%、1.6%;在加熱回流提取時加樣回收率分別為98.2%、97.8%、100.3%、101.4%、99.2%、98.6%、98.4%,對應的RSD 分別為1.7%、1.4%、1.8%、1.9%、1.3%、2.0%、1.5%。
2.7 數據分析
利用Excel 2010和SPSS 23.0對數據進行統計分析,結果采用()表示。采用t檢驗比較兩組間顯著性。運用GraphPad 8.0 和Origin 2019 進行作圖分析。
3 結果與分析
3.1 7個綠原酸類成分在水和乙醇中的溶解度差異
本研究測定了7 個綠原酸類化合物在純水、乙醇2 種常見溶劑中的溶解度(表3)。結果表明,除4-CQA 在2種溶劑中均有較好的溶解性外(溶解度>150 mg·mL-1),其余6 個綠原酸類成分經充分振搖并超聲60 min 后,在水中能較快達到飽和狀態,在乙醇中溶解度更大。以CA 為例,純水中溶解度僅為0.28 mg·mL-1,而在乙醇中溶解度高達52.99 mg·mL-1,與ChemSpider 數 據 庫(http://www.chemspider.com/)收載的CA 在乙醇中的溶解度(50 mg·mL-1)基本吻合;3-CQA、3,4-diCQA 和3,5-diCQA在乙醇中即使溶解度接近200 mg·mL-1時,也并未達到溶解平衡,但其在乙醇中的溶解度遠大于純水。7 個綠原酸類成分在乙醇中溶解度是純水中的數十倍甚至數百倍,這和數據庫中列出的溶解性一致,表明乙醇與其自身的極性更為接近。

表3 7個綠原酸類成分在水和乙醇中的溶解度比較 mg·mL-1
3.2 水和乙醇2 種溶劑超聲提取3 種中藥材中綠原酸類成分含量的比較
以純水和乙醇為提取溶劑,利用現代超聲輔助提取法分別提取艾葉、菊花和金銀花中的主要綠原酸類成分,比較3種中藥材中綠原酸類成分在2種溶劑中提取含量的差異。結果表明,3種中藥材不同來源樣品提取含量變化趨勢一致,但不同植物間各綠原酸類成分在2種提取溶劑中含量變化趨勢有明顯差異。在菊花、金銀花樣品中均未檢測到CA(圖2)。艾葉和菊花樣品中各綠原酸類成分均表現為在乙醇中的提取含量低于純水,且2 種植物中5-CQA、3-CQA、CA 和4-CQA 在乙醇中的提取含量接近于0;菊花樣品中3,4-diCQA 在乙醇中的提取含量也接近于0。這與綠原酸類成分對照品在2 種溶劑中的溶解性完全相反,不符合“相似相溶”原理。與菊科植物不同的是,忍冬科植物金銀花樣品中5-CQA、3-CQA 和4-CQA 在乙醇中的提取含量僅略低于純水,差異無統計學意義;而3,4-diCQA、綠原酸A、綠原酸C 的提取含量表現為在乙醇中顯著高于純水,說明金銀花在超聲提取過程中僅部分綠原酸類成分存在“逆溶解度”現象。上述結果表明,中藥材在超聲提取過程中存在“逆溶解度”現象,而不同植物間“逆溶解度”現象強弱不同,這為今后中藥材中綠原酸類成分的提取提供新的思路。
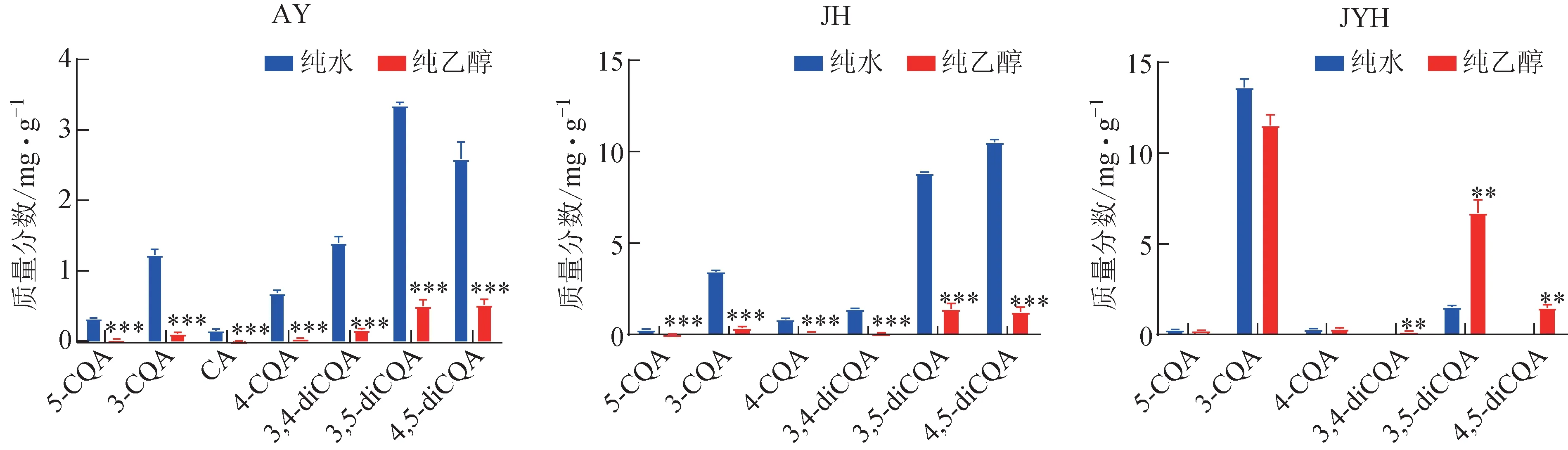
圖2 純水和純乙醇超聲提取艾葉、菊花、金銀花藥材中綠原酸類成分質量分數的差異(, n=3)
3.3 水和乙醇2 種溶劑加熱回流提取3 種中藥材中綠原酸類成分含量的比較
采用加熱回流提取法進一步比較了3 種中藥材在純水、乙醇2 種溶劑中的提取含量差異,結果見圖3。在菊花、金銀花加熱回流提取樣品中也未檢測到CA,不同來源樣品提取規律一致,而菊科植物中各綠原酸類成分在2種溶劑中的提取規律與金銀花有顯著差異。菊科植物在加熱回流時中除3,5-diCQA在乙醇中的提取含量顯著(P<0.05,P<0.01)高于純水外,其余綠原酸類成分在乙醇中的提取率均顯著低于純水,與超聲輔助提取樣品中提取規律基本一致,也存在顯著的“逆溶解度”現象。然而,忍冬科植物金銀花在加熱回流提取中,3-CQA、3,5-diCQA和4,5-diCQA 在乙醇中的提取含量表現為顯著高于純水,呈現“逆溶解度”現象的綠原酸類成分包括5-CQA、4-CQA和3,4-diCQA。該結果表明中藥材在加熱回流提取過程中也存在“逆溶解度”現象,同樣以艾葉、菊花更為顯著。
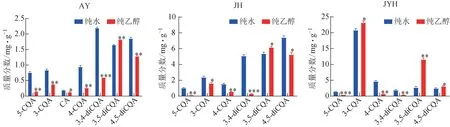
圖3 純水和純乙醇加熱回流提取艾葉、菊花、金銀花藥材中綠原酸類成分質量分數的差異(, n=3)
3.4 金銀花中綠原酸類成分在不同體積分數乙醇中的提取規律比較
對金銀花中5-CQA、3-CQA、4-CQA、3,4-diCQA、3,5-diCQA 及4,5-diCQA 6 個綠原酸類成分在不同體積分數乙醇(0、25%、50%、75%、100%)中的提取含量進行分析,結果見圖4。金銀花樣品中6個綠原酸類成分都是隨乙醇體積分數的升高呈現先增高后降低的趨勢,均在乙醇體積分數為50%時有最高的提取率。根據“相似相溶”原理,50%體積分數乙醇的極性與金銀花樣品中6 個綠原酸類成分的極性相當。該結果與先前的報道基本一致[12],當乙醇體積分數>50%后,醇溶性成分溶出增加,影響了酚酸類物質的提取。
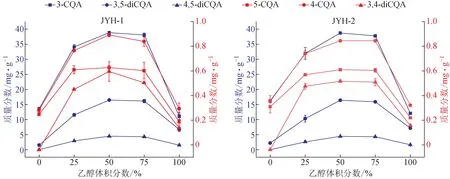
圖4 不同體積分數乙醇提取金銀花樣品中綠原酸類成分的提取規律(, n=3)
3.5 艾葉中綠原酸類成分在不同體積分數乙醇中的提取規律比較
以不同體積分數乙醇提取4份不同產地艾葉樣品中綠原酸類成分,共檢測到新綠原酸、3-CQA、CA、4-CQA、3,4-diCQA、3,5-diCQA、4,5-diCQA 7 個綠原酸類成分。結果顯示,不同艾葉樣品中7個綠原酸類成分含量隨乙醇體積分數變化規律一致(圖5),即隨著乙醇體積分數的升高,3,4-diCQA、3,5-diCQA和4,5-diCQA的含量呈現先升高后降低的趨勢,在乙醇體積分數為50%時提取含量最高,與金銀花變化趨勢相同;而5-CQA、3-CQA、CA和4-CQA的含量則是隨著乙醇體積分數的升高而逐漸減少,在純水中的提取含量最高,呈現顯著的“逆溶解度”現象。結合金銀花樣品在不同體積分數乙醇中的提取規律,說明綠原酸類成分在不同植物中提取規律有顯著差異。因此,在中藥材提取過程中應先探索目標成分在不同溶劑中的提取率以確保較高的得率。
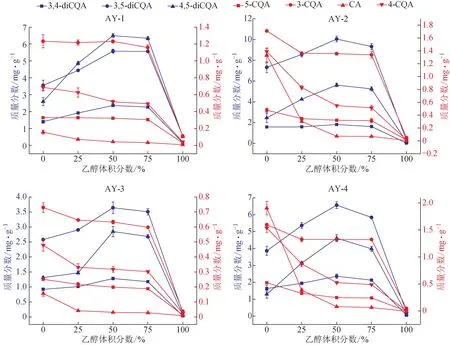
圖5 不同體積分數乙醇提取艾葉樣品中綠原酸類成分的提取規律(, n=3)
3.6 菊花中綠原酸類成分在不同體積分數乙醇中的提取規律比較
在4份不同來源菊花樣品中檢測到5-CQA、3-CQA、4-CQA、3,4-diCQA、3,5-diCQA、4,5-diCQA 6 個綠原酸類成分,未檢測到CA。不同菊花樣品中綠原酸類成分含量在不同體積分數乙醇中變化規律保持一致(圖6)。菊花在提取過程中雖“逆溶解度”現象明顯,但不同于艾葉的是,菊花樣品中隨乙醇體積分數的升高含量逐漸降低的是5-CQA、4-CQA和3,4-diCQA,3個綠原酸類成分在水中含量最高;而含量呈現先增高后降低的成分包括3-CQA、3,5-diCQA、4,5-diCQA,在50%體積分數乙醇下提取含量最高。上述結果表明,中藥材提取過程中綠原酸類成分的提取規律即使在同科不同屬種植物間也呈現顯著差異。
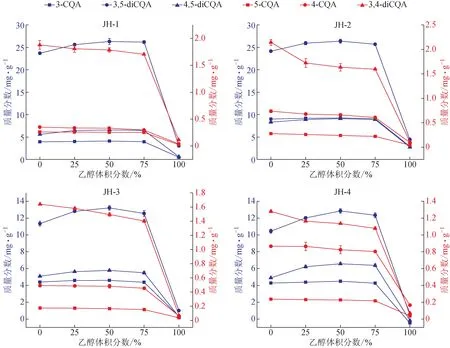
圖6 不同體積分數乙醇提取菊花樣品中綠原酸類成分的提取規律(, n=3)
4 討論
中藥材中化學成分多樣、關系復雜,探究其有效成分在不同條件下的提取規律及相互作用,對優化提取工藝及深入挖掘應用價值具有重要意義[13-14]。本研究基于UHPLC,測定了7 個綠原酸類成分在純水、乙醇中的溶解性能,發現幾種綠原酸類成分在乙醇中的溶解度是純水中的數倍甚至數十倍、數百倍,基本符合其微溶于冷水、易溶于乙醇的性質。而在對菊科植物艾葉、菊花和忍冬科植物金銀花3種常見大宗中藥材進行綠原酸類成分提取過程中發現,2 種菊科植物中綠原酸類成分在超聲提取時乙醇的提取含量顯著低于純水,與各成分單體溶解度完全相反;加熱回流提取時除3,5-diCQA 外,其余綠原酸類成分呈現相同的現象。忍冬科植物金銀花在提取時也有部分綠原酸類成分呈現該現象,超聲提取時5-CQA、3-CQA 和4-CQA 在乙醇中的提取率略低于純水;加熱回流提取時5-CQA、4-CQA 及3,4-diCQA 在乙醇中含量顯著低于純水。說明該現象在中藥材提取過程中普遍存在,但可能因中藥種類、提取方法而異,如不同溶劑對不同植物組織細胞破壞程度不同,進而影響成分的提取率。因此,今后在中藥材綠原酸類成分的提取過程中應將“逆溶解度”現象考慮在內,從而選擇合適的提取條件及提取溶劑以獲得更高的提取率。
為進一步揭示中藥中綠原酸類成分提取中的“逆溶解度”現象,本研究以超聲提取探究了3 種中藥材中綠原酸類成分在不同體積分數乙醇(0、25%、50%、75%、100%)中提取規律。結果顯示,金銀花中檢測到的6 個綠原酸類成分的含量隨乙醇體積分數的增大先增大后減小,并且均在乙醇體積分數為50%時達到最高值。當乙醇體積分數>50%,各綠原酸類成分提取含量逐漸降低,從而呈現出“逆溶解度”現象,推測可能是植物中醇溶性雜質、色素等親脂性強的成分過多阻礙了綠原酸類成分的溶出[15]。這與一些鹽類在水等溶劑中的溶解度隨著溫度的升高而增加,當溫度超過某一數值后,其溶解度隨著溫度的增加反而下降的“逆溶解度”特性相似。然而,在艾葉和菊花中卻表現出更為顯著的“逆溶解度”現象。艾葉中3 個異綠原酸成分變化趨勢與金銀花一致,但5-CQA、3-CQA、CA和4-CQA卻在純水中呈現最高的提取率,并隨著乙醇體積分數的升高而降低。菊花與艾葉略有不同的是,綠原酸含量變化趨勢與金銀花相同,而3,4-diCQA 在純水中提取的含量最高。
目前,呈現出上述提取規律公認的原因主要是成分溶解率受提取過程中溶劑對細胞的滲透性能、藥材中各組分相互影響與共溶、分子在細胞內存在狀態等影響。然而,本研究認為,植物中不同綠原酸類成分間可能還存在某種特異的結合方式,且在不同植物間也略有差異。有研究表明,中藥復方水煎過程中有效成分可產生相互作用,肉桂酸和黃連素自組裝形成超分子聚合物,從而明顯增強對臨床多重耐藥金黃色葡萄球菌的抑制作用[16]。由相同或不同種類的單體通過非共價鍵自組裝形成的超分子聚合物,基于非共價鍵的可逆性可區別于單體,從而顯示出獨特的特性[17]。利用中藥化學成分的分子識別與自組裝特性,總結出各類成分提取規律,開發新的提取、分離方法,不僅能兼顧中藥的多成分群,更能提高中藥提取分離的特異性、準確性、穩定性[18]。然而,“逆溶解度”現象中存在的相互作用還需進一步深入研究其內在機制才能給出合理的解釋。
綜上所述,本研究揭示了中藥材中綠原酸類成分在不同植物、不同提取方法、不同提取溶劑中存在強弱不一的“逆溶解度”現象,為綠原酸類成分的提取和深入研究提供了新的思路,對中藥配方顆粒制備工藝優化、植物化學分離條件優選及工業生產有重要的指導意義。
