富鋰錳基正極材料研究進展
2022-06-27 13:18:30李俊瀟李天樂孟紹良趙久成吳軍王文舉
能源研究與利用 2022年3期
李俊瀟,李天樂,孟紹良,趙久成,吳軍,王文舉
(1.南京理工大學能源與動力工程學院,南京 210094;2.南京瑞華新能源電池科技有限公司,南京 210037)
如今,工業持續高速發展,伴隨其中的能源危機和環境污染問題已經無法再容忽視。傳統燃油為動力源的汽車不但會使能源危機進一步加重,而且其排放的尾氣中有大量的有害氣體,這無疑會加重環境的污染,而新能源汽車的出現能夠有效緩解能源危機和環境污染這兩大問題。新能源汽車的核心是電池,隨著鋰離子電池的發展和近年來國家政策的扶持,動力鋰離子電池已經成為了新能源汽車的不二之選。作為一種電能儲能器件,鋰離子電池無論是在新能源汽車還是非并網的新能源發電側,都可以很好地實現電能的轉移[1]。同時,鋰離子電池還對移動電子器件等新興領域的發展至關重要。
最早的商業化鋰離子電池是由索尼公司在1991年推出的,當時是使用金屬氧化物基正極和軟碳負極。經過30多年的發展,鋰離子電池的正極和負極材料已有多種選擇,目前投入商業化使用的正極材料主要有錳酸鋰(LiMn2O4)、磷酸鐵鋰(LiFePO4)、鈷酸鋰(LiCoO2)、鎳鈷錳酸鋰(LiNixCoyMn1-x-yO2)及鎳鈷鋁酸鋰(LiNixCoyAl1-x-yO2)。當前大規模投入使用的正極材料在實際應用中比容量都在110~200 mA·h/g之間,但已實現商業化的負極材料的比容量卻很高,約是正極材料比容量的3~20倍不等,石墨負極比容量為372 mA·h/g,硅基負極比容量更是高達2 000 mA·h/g。正極材料正在成為鋰離子電池性能進一步提升的瓶頸。在已知正極材料中,富鋰錳基層狀氧化物(xLi2MnO3·(1-x)LiMnO2)的比容量高達250 mA·h/g,被認為將是提升鋰離子電池能量密度的理想選擇。但富鋰錳基材料也有很多缺陷,如首次不可逆容量高、循環和倍率性能差、電壓衰減明顯和安全性能差等,因此相關學者做了很多富鋰錳基材料改性方法的研究。幾種典型正極材料的電化學特征見表1。
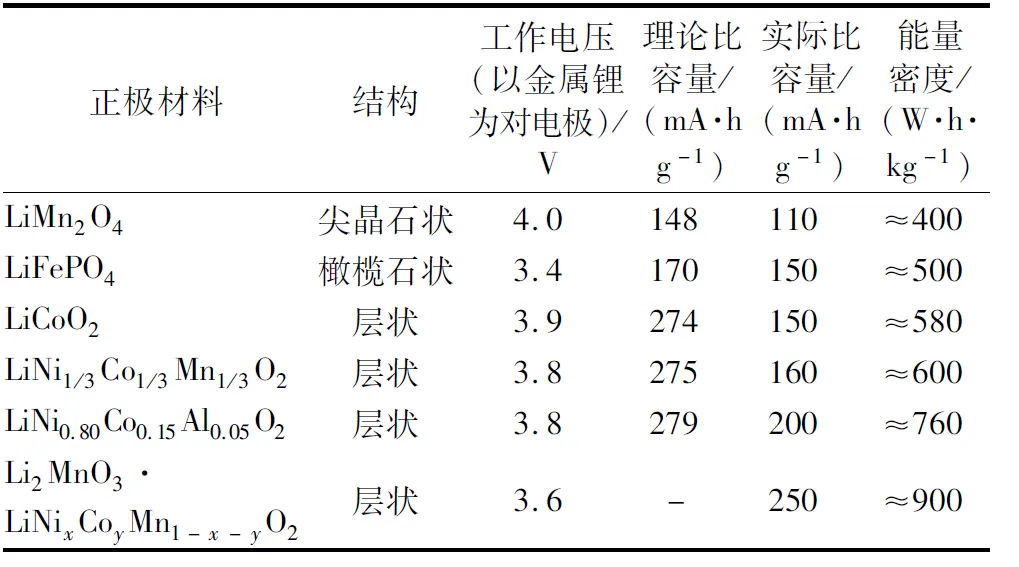
表1 幾種典型正極材料的電化學特征
1 鋰離子電池充放電機理
鋰離子電池主要由正極材料、負極材料、電解質和隔膜這四個部分組成。其中正負極為能量的載體,這是鋰離子能夠在其中進行可重復的游離和結合的保障;電解質是鋰離子可以在電解液中自由遷徙并降低自放電率的保障;而隔膜則是為了把電池正負極分隔開,只允許鋰離子通過。鋰離子電池的工作依賴于鋰離子在正負極濃度的不同帶來的鋰離子的轉移。充電時,鋰離子從負極轉移到正極,正極變為富鋰態。充放電過程中,隨著鋰離子的轉移,相同數量的電子同時在外電路中移動,使正負極發生氧化還原反應。可充電鋰離子電池的工作原理圖(以LiCoO2電池為例)[2]如圖1所示。
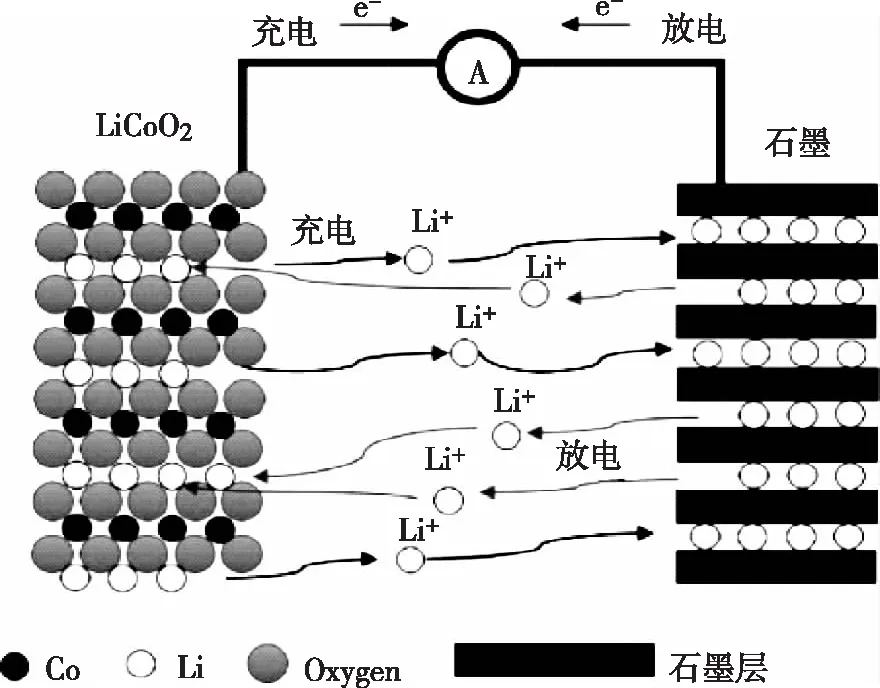
圖1 可充電鋰離子電池的工作原理圖
2 富鋰錳基正極材料
多年來研究人員一直致力于追求更高性能的電池體系,通過當前達成的成果可以看出,電池里非活性物質的占比已經非常低了,以往我們通過降低非活性物質的量來提高電池能量密度的方法已經沒有太多發揮空間了。想要提高鋰離子電池的能量密度,最直接的方法就是提高正負極材料的能量密度,即使用更高能量密度的電極材料。正負極的容量適配是設計鋰離子電池的首要準則。而目前商用的鋰離子電池正極材料的比容量普遍偏低,因此需要更重的正極材料才能匹配相應比容量的負極(至少2 g的正極材料才能匹配1 g的石墨負極材料),碳硅負極材料需要的正極材料往往更多。所以我們要積極尋找新一代高容量正極材料來取代傳統正極材料。
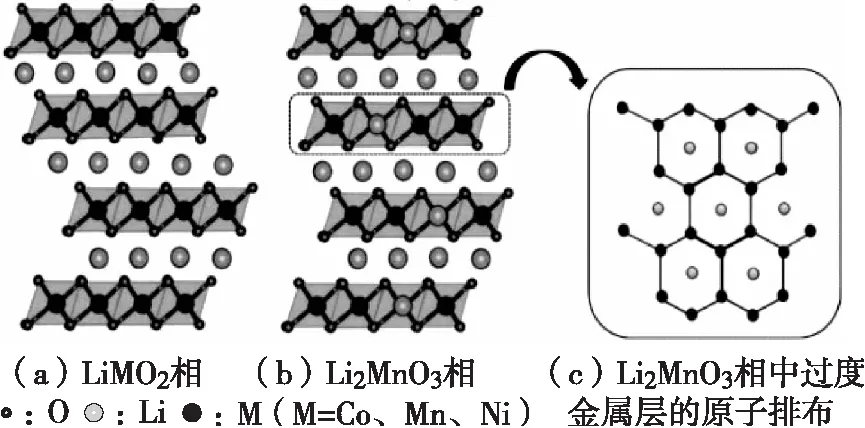
圖2 富鋰錳基正極材料結構示意圖
富鋰錳基正極材料放電比容量達250 mA·h/g以上,幾乎是目前大規模投入量產使用的正極材料可用容量的兩倍左右;富鋰錳基以錳元素為主,貴重金屬含量少,錳元素價格低,因此富鋰錳基整體價格較低,富鋰錳基的結構示意圖如圖2所示[3]。同時,相比于常見的三元鈷酸鋰和鎳鈷錳材料,這種材料兼具成本跟安全性的優勢。所以,在實現下一代鋰電池突破400 W·h/kg,甚至500 W·h/kg的目標途中,富鋰錳基正極材料是理想的選擇也是技術的關鍵。富鋰錳基正極材料雖然有很多優點,但其也有實際應用中不容忽視的缺陷。盡管近年來國內外在其研究方面取得了重要進展,但若想實現富鋰錳基正極材料的商業化,還要面臨幾個挑戰:(1)首次充放電的可逆容量損失大和庫侖效率低;(2)容量及電壓衰減嚴重;(3)倍率性能差;(4)安全性能差。
2.1 富鋰錳基正極材料的缺陷
2.1.1 首次充放電的可逆容量損失大和庫侖效率低
由于富鋰正極材料xLi2MnO3·(1-x)LiMO2晶體構造的特殊性,致使其充放電過程中,存在首次不可逆容量損失較大,首圈庫倫效率較低的問題,圖3為不同組分富鋰錳基電池首次充放電曲線[4-5],和后續循環過程的充放電曲線有明顯差異。在電化學反應中,富鋰錳基正極材料中Li2MnO3容易被激活,晶格氧也會發生流失,從而導致其表面構造和組分的變化,進而出現一些問題。圖3中呈現出兩個明顯的充電平臺,在4.5 V前的平臺出現來源于其中的Li-MO2發生鋰離子脫嵌現象,Ni2+和Co3+被氧化;第二次平臺出現在4.5 V附近,但該平臺只在首次充電時存在。研究認為,當第一次充電電壓高于4.5 V后,不活躍的Li2MnO3組分被活化并參加進反應中,晶格中的O2-通過“Li2O”的形式從Li2MnO3組分中溢出,剩下的部分則成了新組分MnO2。首次充電活化Li2MnO3相會產生含鋰化合物且此過程只能單向進行,導致第一次充電損耗容量非常大,進而導致首次庫倫效率過低[6]。另外,充電時正極材料也會跟電解液中的Li+相互反應,反應會形成SEI膜,特別是當電壓高于4.5 V時會提高該反應的不可逆性,導致Li+損耗增多,這也是引起材料第一次充放電庫倫效率降低的因素之一。
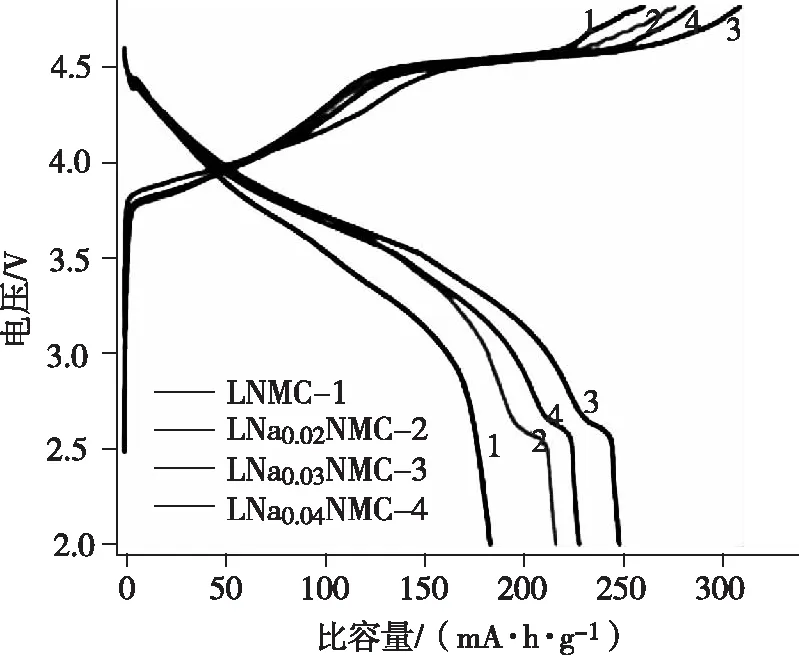
圖3 不同組分富鋰錳基電池首次充放電曲線
2.1.2 容量及電壓衰減嚴重
富鋰錳基正極材料最大的問題就是電壓及容量衰減。電壓衰減的定義是材料在經過多次充放電后活動電壓有明顯下降的跡象,活動電壓的降低還會很大程度的縮減材料能量密度。圖4是不同富鋰錳基材料的比容量隨循環圈數的變化情況。富鋰錳基材料的電壓衰減是因為其體相結構發生了變化,充電時材料會失去表面的晶格氧,材料中的過渡金屬離子由于氧的配位數減少而發生遷移,晶體由層狀結構變為尖晶石結構,最終又變成無序巖鹽結構。晶格氧的失去導致了內部的Ni和Mn過渡金屬元素向表面移動,因此材料表層呈現出缺Li、多Mn和Ni的尖晶石相重構層。另外,逃逸的氧會和電解液發生反應,隨著反應次數的不斷增多,晶格氧與電解液的反應越來越劇烈,從而導致尖晶石相陸續由材料表面向中部延申,由此加劇了材料電壓和容量衰減問題[7]。大量實驗表明以下兩點是引起電壓衰減的主要因素:(1)材料的自身結構的轉變,
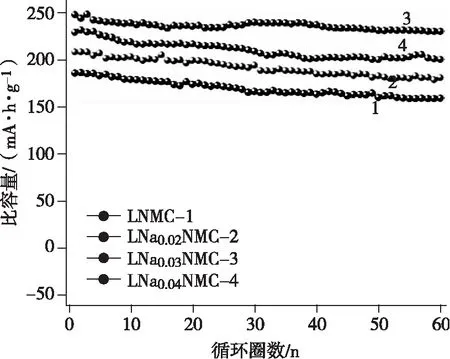
圖4 富鋰錳基正極材料的循環性能
由層狀變為尖晶石相。(2)電極材料與電解液接觸的地方發生惡化以及發生副反應,使電極表面形成SEI膜致使電極極化的增大。除此之外還存在其他的次要原因。劉彥辰等[8]還介紹了幾種影響電壓衰減的因素,分別是過渡金屬元素對電壓衰減的影響、氧氣的不可逆釋放對電壓衰減的影響和測試條件對電壓衰減的影響。
2.1.3 倍率性能差
圖5是不同倍率下富鋰錳基正極材料的倍率性能,從圖5中可以發現倍率越高,材料的比容量越低。分析富鋰錳基材料的循環過程發現,首次循環后,其中的一些材料以LiMnO2結構加入到氧化還原反應中,因此穩定性能不佳。同時材料的離子擴散能力多數偏低,因此導致倍率性能也相對較差[9]。另外,電極材料和電解液的共存,讓材料表面的晶格氧更加活躍,氧的脫出一方面會使材料比容量減小,另一方面氧還會作用于電解液減少其中的可逆Li+,反應生成不可逆的化合物累積在電極表面,妨礙 Li+從電極材料表面向內部的遷移,直接導致富鋰錳基正極材料的倍率性能較差。
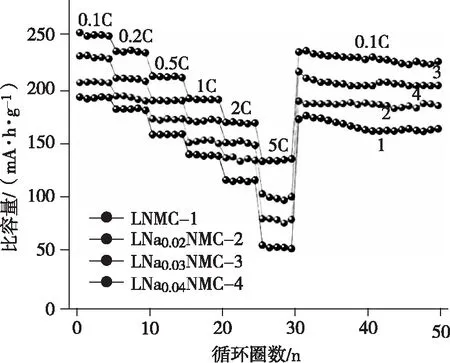
圖5 不同倍率下富鋰錳基正極材料的倍率性能
2.1.4安全性能差
富鋰錳基材料在安全方面也存在一定的隱患,具體表現為首次充電時會有氧氣的析出,氧氣析出后材料會進一步與電解液發生反應,最終導致產氣問題[9]。
2.2 富鋰錳基正極材料的制備方法
采用不同方法合成的富鋰錳基材料在微觀構造、微粒直徑、聚團水平以及比表面積等方面都會有所不同,材料的電化學性能也會受到影響。材料制作的工業操作難度、投資成本也會影響到富鋰錳基材料的實際生產應用。因此,我們需要不斷改良以尋求最佳的合成方法,目前比較主流的制備的方法有溶膠凝膠法、高溫固相法和共沉淀法。
2.2.1 溶膠凝膠法
溶膠凝膠法是在溶液中將過渡金屬離子充分混合,再加入絡合劑使其形成溶膠,然后蒸發溶劑后得到凝膠,對凝膠進行干燥和煅燒即得到成品。溶膠凝膠法的優點是制得的材料元素充分混合、粒徑分布窄、顆粒粒徑小,缺點是操作流程復雜,大批量生產難度較高,難以實現工業化[10]。
2.2.2 高溫固相法
高溫固相法是含有不同金屬元素的化合物機械混合后,在700~1 000 ℃下煅燒,最終得到產物。該方法的優點是操作簡單,便于工業化生產,缺點是得到的產物圍觀分布不均勻,電化學性質不穩定。邱家欣[11]對傳統高溫固相制備工藝加以改良,分別以有機物聚乙烯吡咯烷酮(PVP)作為分散劑,以有機物乙二醇(EG)作為粒度調節劑來進行沉淀反應,同時利用水熱反應輔助制出MnO2中間體,將其作為錳源制備微米或者納米結構的富鋰錳基正極材料。改良后的方法不僅與傳統的高溫固相法一樣操作簡單,而且還改善了制備材料的晶粒團聚問題,對材料形貌的控制也更為精確。
2.2.3 共沉淀法
共沉淀法是采用某種絡合劑,使溶液中的過渡金屬離子先與絡合劑絡合,再與沉淀劑反應形成沉淀。該方法需要嚴格的調控PH值,制得的樣品具有優異的形貌結構,多為均勻大小的規則球體二次顆粒。目前工業生產中多采用此方法。李文明等[12]使用草酸鹽共沉淀方法,配合后續混鋰焙燒制備了鋰離子電池0.5Li2MnO3·0.5LiCo0.5Mn0.5O2富鋰錳基正極材料。研究材料的化學性能,可以看出在低攪拌轉速條件下制作的球狀顆粒樣品相比于其他樣品有更高的振實密度和更優異的電化學性能。
2.3 富鋰錳基正極材料的改性方法
2.3.1 組分研究
富鋰錳基材料的首次發現是KIM等[13]以不具有電化學活性的層狀材料Li2MnO3穩定擁有活潑電化學活性的材料LiMnO2,從而構成富鋰層狀正極材料xLi2MnO3·(1-x)LiMnO2。將這種正極材料充電到4.5 V后,材料將被活化,其比容量將高達280 mAh/g[14]。富鋰錳基正極材料的結構相當復雜,主要是由于x值的不同,為了獲得滿足不同需求的各種性能,通常采取改變x從而改變材料結構進而改變材料性能的辦法。PECHEN等[15]利用共沉淀法合成了xLi2MnO3·(1-x)LiMn1/3Ni1/3Co1/3O2(x=0.2,0.35,0.5)材料,并比對了不同x值對材料的相結構、微觀形貌、粒徑分布、密度和電化學性能的影響,對電化學測試結果進行分析,當x=0.35時兩相的組合能使材料呈現出最優異的電化學性能,經過110次充放電循環容量保持率高達85%。
2.3.2 摻雜
為了提升材料的首次庫侖效率,我們通常對富鋰錳基材料進行離子摻雜,這樣做可以提高材料的循環穩定性,從而削弱循環過程中可能會出現的由于金屬陽離子混排導致的材料結構問題[9]。目前主要的離子摻雜形式有:陽離子摻雜、陰離子摻雜、聚陰離子摻雜、共摻雜。忽小宇等[16]通過在前驅體和鋰源的高溫固相反應階段加入五氧化二釩作為摻雜劑制備了釩摻雜的富鋰錳基正極材料(LNCM)。對這兩種正極材料進行表征和電化學測試,結果表明,釩摻雜后,材料的電荷轉移阻抗明顯降低,倍率性能也得到有效的改善。
2.3.3 表面修飾
體相摻雜雖然可以降低電荷轉移阻抗和提高鋰離子擴散能力,但是并不能避免材料與電解液的接觸。對材料表面進行包裹可以把材料保護起來,使材料免于與電解液直接接觸,從而減小材料被電解液腐蝕的風險,達到改善材料的電化學性能的效果。目前常用的包裹材料為惰性氧化物、磷酸鹽、氟化物、含鋰化合物及導電聚合物等。合肥國軒高科動力能源有限公司發明了一種鉬摻雜氧化鋅包覆富鋰錳基正極材料的制備方法[17],有效的隔絕電極與電解液的接觸,改善了電極材料與電解液間的導電能力及離子傳輸速率,從而提高了富鋰錳基正極材料的容量保持率及循環性能。
3 結語
富鋰錳基正極材料具有高比容量,低成本等優勢,放電比容量達250 mA·h/g以上,將是未來鋰電池達到400 W·h/kg,甚至500 W·h/kg的技術關鍵,具有非常高的應用前景。但如果不解決以下幾個技術方面的關鍵問題,富鋰錳基材料將無法在鋰離子電池上實際運用:一是降低首次不可逆容量損失;二是改善倍率性能和提高循環壽命;三是削弱循環過程的電壓衰減;四是提高安全性能。
目前已有多種手段來解決這些問題:改變組分結構、包覆、酸處理、摻雜、預循環、熱處理等。(1)通過合理的改變富鋰錳基正極材料的組分和結構可以提高電池的性能,但該研究技術目前還沒有完備的理論支撐,目前我們所作的相關研究較少,得到的結果具有一定的局限性;(2)通過對富鋰錳基材料進行摻雜可以顯著提高鋰離子電池的倍率性能,目前已有很多研究者做了用不同離子摻雜富鋰錳基材料的實驗;(3)通過對富鋰錳基材料進行表面包覆可以在材料表面形成保護層,能改善鋰離子電池的容量保持率及循環性能,目前已發現多種材料都可用于富鋰錳基材料的表面修飾。但是上面這些方法都只針對單個方面提升材料的性能,還沒有萬全之策。期望未來我們能把多種改性辦法結合起來,找到綜合提升富鋰錳基性能的最佳改性方案。
