Anti-nuclear matrix protein 2+ juvenile dermatomyositis with severe skin ulcer and infection:A case report and literature review
2022-06-23 02:06:56YaTingWangYuZhangTaoTangChongLuoMingYueLiuLiXuLiWangXueMeiTang
World Journal of Clinical Cases 2022年11期
lNTRODUCTlON
Juvenile dermatomyositis (JDM) is a rare systemic autoimmune disease characterized by vascular disease that mainly affects muscles and skin, as well as the lungs, intestines, heart, and other organs[1-3]. JDM can also lead to macrophage activation syndrome, which is a potentially fatal complication of a number of rheumatological conditions[4]. JDM is the most common inflammatory myopathy in children and has been reported to affect 1.9 individuals per million children in the United Kingdom and 2.4-4.1 individuals per million children in the United States[5,6]. Skin ulcers are one of the severe manifestations of childhood dermatomyositis; however, cases of severe skin ulcers with infections are rarely reported. Here, we report a single case of a 2-year-old female patient who suffered from JDM and whose myositis-specific autoantibodies (MSAs) were positive for anti-nuclear matrix protein 2 antibody, with progressively worsening skin ulcers and severe infections.
CASE PRESENTATlON
Chief complaints
A 2-year-old Chinese girl came to the Department of Rheumatology and Immunology with a heliotrope rash for 1 mo and muscle weakness for 10 d.
She walked boldly up to the door and entered the room as if the whole place belonged to her, and quite frightened the poor girl, who was startled at the sight of her old enemy
History of present illness
This patient developed a heliotrope rash, periorbital edema, and nailfold capillary changes within 1 mo,with symmetric proximal muscle weakness. There was no fever, cough, hoarseness, or sensory disturbance.
In European folklore, each of the four winds has a different personality. The gentle East Wind brings warmth and rain. The vigorous West Wind brings dry weather. The South Wind brings heat and drought. The North Wind is the strongest of the four and brings winter and bitter cold to Northern Europe. (Jobes 1961, 1682-1683). The genders111 of the winds are malleable112 and often not designated as they are in this tale.
History of past illness
When the patient was diagnosed with anti-nuclear matrix protein 2 (NXP2)+ juvenile dermatomyositis,she initially received intravenous immunoglobulin (IVIG; 1 g/kg) for 2 d and high-dose glucocorticoids(GC; 15 mg/kg) for 3 d, after which she was treated with oral methylprednisolone (1.15 mg/kg.d),intravenous cyclophosphamide (IV CYC; 0.1 g/kg) for 2 d, methotrexate (MTX; 7.5 mg qw), hydroxychloroquine (0.05 g qd), and other symptomatic and supportive treatment. Following these treatments,the child's rash and edema basically disappeared, and her muscle strength improved.
Personal and family history
Persistent progression of skin ulcers has also been associated with neglected assessment of skin manifestations and severity of JDM. An expert group recommends that the follow-up of patients with JDM should focus more on the evaluation of the skin, including the use of multiple scales[8].The Cutaneous Dermatomyositis Disease Activity and Severity Index(CDASI) and the Cutaneous Assessment Tool (CAT)have good interrater reliability and correlation with other measures of activity and damage in children with JDM[15]. The child in our case had prominent skin damage in the later stage. Early assessment of skin may help with follow-up treatment. Additional assessment tools can be used in clinical practice to more comprehensively assess disease activity[16].
Physical examination
After admission, the patient’s weight was 14 kg. There were heliotrope rashes on her face, periorbital edema, changes in nailfold capillaries, Gottron papules on the dorsal surface of the proximal interdigital(PIP), symmetric proximal muscle weakness of arms and legs, no erythema butterfly, and arthritis.
Laboratory examinations
Investigations revealed elevated creatine kinase of 12647 U/L (reference range, 50-220 U/L), lactate dehydrogenase (LDH) of 1358 U/L (reference range, 80-300 U/L), erythrocyte sedimentation rate (ESR)of 79 mm/h (reference range, 0-20 mm/h), and ferritin of 726 ng/mL (reference range, 10-120 ng/mL).The autoantibody, immunoglobulin, and complement profiles were normal. Anti-nuclear matrix protein 2 (NXP-2) antibody was positive in the myositis spectrum, as determined by the dot-ELISA method, and no other myositis-associated autoantibodies were present (Table 1).
Imaging examinations
Magnetic resonance imaging (MRI) of the bilateral thighs revealed inflammatory changes in the musculature and subcutaneous fat layer of the thigh muscles on both sides, which were also characteristic of dermatomyositis. Electromyography (EMG) showed that the motor unit potential amplitude of the tibialis anterior and rectus femoris muscle was reduced, and the duration was shortened, thus suggesting myogenic abnormalities.
FlNAL DlAGNOSlS
TREATMENT
The patient had no significant medical or surgical history.
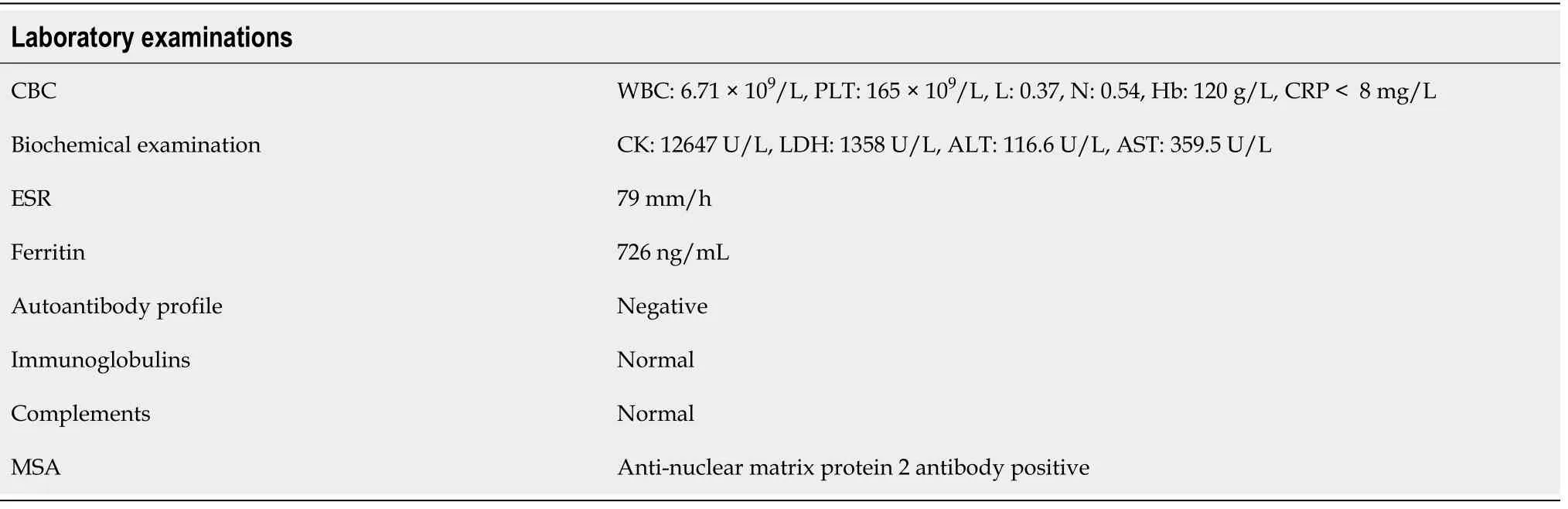
In the following 8 mo, the girl was hospitalized 9 times, and she received IVIG 9 times (1 g/kg for 2 d each time) and IV CYC 6 times, replacing MTX and hydroxychloroquine with mycophenolate mofetil(MMF) and thalidomide. From the 4th month of the illness, she was treated with tofacitinib (2.5 mg bid).In the first three months after diagnosis, the child developed livedo reticularis on the skin of the extremities (Figure 1A) and ulcers on the buttock and left upper arm, while the ulcerated surfaces gradually increased. In the 6month, yellow necrotic fascia was visible on the left buttock (Figure 1B).After she received proper wound care, anti-infection, and adjustment of immunosuppressants, the ulcers improved (in the 8month), showing gradual scabbing. Nonetheless, her mother discontinued the patient’s hospitalization. One month later, the patient suffered fatigue, anorexia, and multiple ulcers on the whole body again, which were worse than before. She never showed calcinosis during the whole course of the disease. After rehospitalization, she was given tocilizumab (12 mg/kg). However, when she received 20% of the infusion, the patient developed irritability, and her heart rate increased; thus,the administration of tocilizumab was stopped.
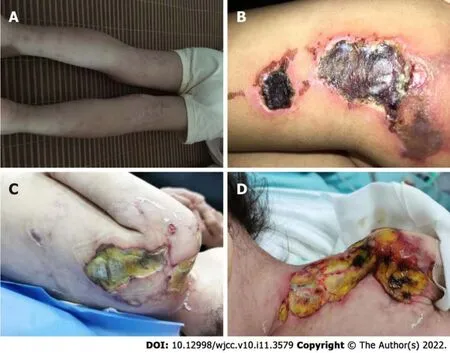
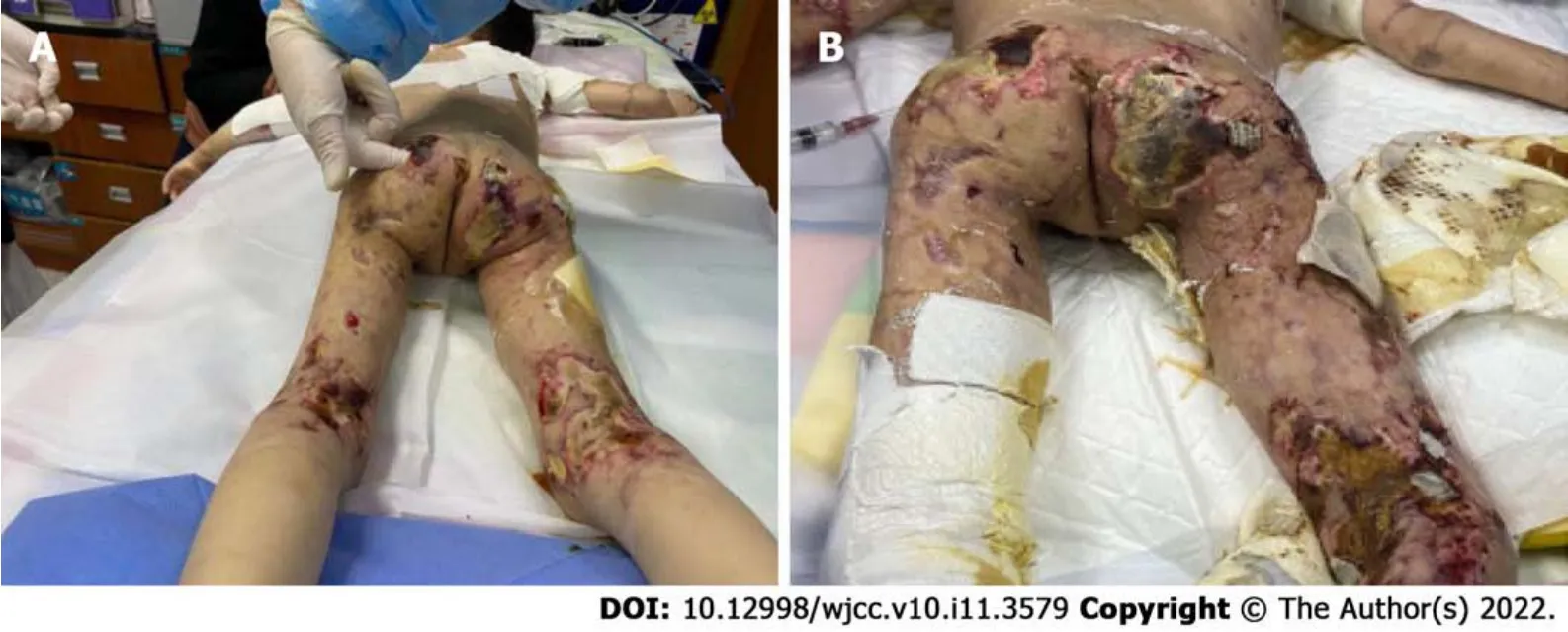
In the 10month after diagnosis, the child was hospitalized for the last time because of bloody stools,fever, anorexia, listlessness, and multiple painful skin ulcers throughout the whole body (Figure 1C and D). The patient could not move on the bed by herself because of low muscle strength. Laboratory examinations revealed CK of 289 U/L, LDH of 689 U/L, and ESR of 92 mm/hr. Skin tissue biopsy of the left upper arm and left neck suggested epidermal necrosis, hyperplasia of subepidermal fibers and fatty tissues, visible vitreous and mucous changes, multifocal necrosis of the subepidermis and dermis, and focal chronic inflammatory cell infiltration. We used ceftazidime to fight infections and added vancomycin after 2 d. Pathogen detection in the pus from the buttock suggested.Accordingly, the antibiotics were adjusted to vancomycin and meropenem; however, the ulcers further deepened (Figure 2A and B). After 9 d, re-examination of the culture revealedand. Based on susceptibility testing, the antibiotics were changed to amikacin and ciprofloxacin. Other treatments included adjusting oral methylprednisolone at 6 mg qd (0.5 mg/kg·d) to intravenous methylprednisolone at 10 mg bid (2 mg/kg·d). During this period, the systemic dressing was changed at least 3 times per week, while IVIG, human albumin, component blood transfusion, and other symptomatic and supportive treatments were intermittently given.
OUTCOME AND FOLLOW-UP
Immediately after the diagnosis of JDM was made, we treated this patient with corticosteroids and immunosuppressants. Systemic corticosteroids are the gold-standard initial treatment for JDM.However, they should not be used as monotherapy because this approach is frequently ineffective and associated with the development of unacceptable long-term adverse effects[26]. Immunosuppressants have steroid protection and are recommended even for mild cases to minimize the adverse effects of long-term GC treatment. Treatments used for refractory disease include IVIG, cyclophosphamide,cyclosporine, azathioprine, MMF, hydroxychloroquine, tacrolimus, rituximab, infliximab, and autologous stem cell transplantation[8]. Some studies suggest that treatment with GC and cyclophosphamide combined with calcineurin inhibitors is very important for dermatomyositis patients with skin ulcers or other severe manifestations[27]. A JAK-inhibitor (JAKi) can be used to treat JDM and has been reported to be partially effective for interstitial lung disease and cutaneous dermatomyositis[28,29].However, in the present case, a JAKi did not stop the progression of the disease. For NXP2+ JDM and severe JDM, many studies have reported a favorable therapeutic effect of rituximab[19,30], which can reduce disease activity and reduce GC use. Additionally, rituximab can ameliorate the skin symptoms of refractory JDM and is effective for skin ulcers of JDM[19]. However, because the skin ulcers in this case were accompanied by severe infection, we were worried that the use of rituximab would further aggravate the infection, so it was not used. We tried tocilizumab, but the patient could not tolerate it.Plasmapheresis can be considered for the treatment of cases refractory to immunosuppressants[8].
I wasn t prepared for a reply, but with resignation, he said, I really am homeless and I really am hungry! You can come with me and watch me eat! But I kept on walking.
DlSCUSSlON
JDM is a rheumatic disease that occurs in childhood, with a mortality rate reaching approximately < 4%,which is second only to systemic lupus erythematosus[7,8]. With early treatment, 30%-50% of patients are likely to achieve remission within 2-3 years from the onset of the disease. In addition to the characteristic skin lesions, other criteria include symmetric proximal muscle weakness, elevated serum muscle enzyme levels, myopathic changes on electromyogram, and typical muscle biopsy results[9].
An ulcer is one of the most serious skin manifestations of JDM and is widely regarded as an indication for more intensive treatment[10]. In different cohorts, the incidence of JDM skin ulcers has been reported to range from 2.6%-23%[7,11,12]. Rare cases have shown such severe skin ulceration with multiple pathogenic infections. Cutaneous ulceration may occur on any soft tissue in JDM, especially the armpits, elbows, or pressure points. Although rare, gluteal ulcers are more likely to worsen due to irritation caused by stool, urine, friction, and maceration, especially in infants and young children[13].In our case, the child’s gluteal ulcer had a protracted condition that gradually expanded and deepened and was accompanied by refractory bacterial infection. Infection and JDM promote each other, which increases the difficulty of treatment and leads to prolonged and unhealed ulcers. A case of JDM in an infant with gluteal ulcer has been reported in Japan. After treatment with high-dose glucocorticoids with cyclophosphamide and MTX, the ulcer gradually healed[13]; however, this case was not accompanied by serious infection. To prevent infection and promote ulcer healing, topical treatments and routine care for skin ulcers are necessary. In 2020, guidelines for skin ulcers related to connective tissue diseases were published in Japan, which in detail described systemic and local medication,nursing, and other treatment methods for skin ulcers and for different connective tissue diseases[14].
As our game progressed through the regulation eighteen holes, it was obvious that Dad, playing quite well by his standards, was hanging on for dear life. And at the very last hole, Mark, a gentle giant of a young man, strode forward none-too-confidently to try to sink a short putt that would win the match. As he stood there concentrating, I wanted him to sink the putt and win, but I was equally prepared to play on to a sudden death if necessary. Agonizingly he hit it, and the ball rolled gently-into the cup! A beam of joy lit up his face, and I felt deep in my heart: That s my boy. I thought, too, of the lines of Betjeman:
The child was born at full term. Her parents and other family members had no family history of autoimmune or other diseases.
In recent years, JDM combined with different MSAs has received extensive clinical attention.Different MSAs have been associated with different clinical phenotypes, prognoses, and risks of associated malignancy. In United States and European cohorts, MSAs were reported to be present in approximately 70% of JDM cases[17,18].
It was the afternoon of December 24, the day before Christmas; and as the newest hygienist in our office, I had to work. The only thing that brightened my day was the beautifully decorated Christmas tree in our waiting room and a gift sent to me by a fellow I was dating—a dozen long-stemmed red roses.
For JDM-related skin ulcers, steroids and immunosuppressants should be first used to control the primary disease. However, if skin ulcers are complicated by infections, this type of treatment may exacerbate the ulcers due to the increased susceptibility of the patient to infection[14].Therefore,in such cases, it is necessary to treat the primary disease and the infection simultaneously. According to the pathogenic examination and drug susceptibility results of the site of infection, appropriate antibacterial drugs were chosen, and the use of IVIG was considered. Due to the refractory nature of cutaneous vasculitis in JDM, case studies suggest that nifedipine, sildenafil, intravenous prostaglandins, and bosentan should be added as early adjuncts[26,31].
I never could hold a pencil the right way. I never could color in the lines. Every ime I would try, my hand would cramp2 up and the letters would come out sloppy3, the lines too dark, and the marker would get all over my hands. Nobody wanted to switch papers with me to grade them because they couldn t read them. Keith could, but he moved away.
On the 48day after admission, gastrointestinal bleeding and shock occurred, after which the girl was transferred to the ICU. She was discharged 6 d later and died a few days after leaving the hospital.
These words would aptly describe the vessel from Spain, for herewas the same luxury, and the same parting thought naturally arose: God grant that we once more may meetIn sweet unclouded peace and joy
NXP2 is a protein involved in transcription and RNA metabolism regulation[19]. The autoantibody was first identified in 1997 in childhood myositis and was considered a key biomarker for the diagnosis of idiopathic inflammatory myopathy[20]. Clinically, anti-NXP2+ JDM often manifests as obvious skin rash, muscle weakness, dysphagia, calcinosis, limb edema, younger age of onset and less remission at 2 years[16]. The incidence of anti-NXP2+ JDM is 20%-25% among JDM cases, making anti-NXP2 a common type of antibody[19]. Two different studies in China have revealed detection rates of anti-NXP2 antibodies in JDM of 30.6% and 20%[21,22]. Albayda[23] found that NXP2+ dermatomyositis with limb weakness and neck muscle weakness were more serious than NXP2- dermatomyositis;accordingly, as our patient showed severe weakness. Another feature of NXP2+ JDM is calcinosis,although this was not present in our case. Early diagnosis and treatment of JDM can prevent the occurrence of calcinosis[24]. As there is no definitive treatment for severe calcinosis, surgical treatment is often necessary; however, there is still the possibility of recurrence[25]. Although most NXP2+ JDM patients are sensitive to GC, some patients are prone to severe and refractory JDM manifestations.
In the present case, after high-dose GC treatment, IVIG, MTX, MMF, hydroxychloroquine, a JAKi,thalidomide, and tocilizumab were given to the patient, and improvement was observed; however, the disease was not completely prevented. Further progression of the disease and the combination of primary disease and severe infection in the later period were fatal. It remains unknown whether the early use of vasodilatory agents and rituximab or plasmapheresis at the proper time could save patients’lives, which future studies should address.
Knowing Ali Baba s poverty, the sister was curious to find out what sort of grain his wife wished to measure, and artfully put some suet at the bottom of the measure
CONCLUSlON
Only scarce reports of JDM with severe ulcers accompanied by infection have been reported. Skin ulcercomplicated and anti-NXP2+ JDM usually represent severe cases, and it is important to actively prevent the occurrence of infection while GC and appropriate immunosuppressive therapy are used. According to the different clinical manifestations and immunological indicators of JDM patients, appropriate assessment tools should be used to comprehensively assess the condition of JDM patients at an early stage, and individualized treatment plans should be customized.
Characteristic skin lesions, proximal muscle weakness, elevated serum muscle enzyme levels, EMG myopathic abnormalities, and changes in muscle MRI findings confirmed the diagnosis of juvenile dermatomyositis.
Tang XM conceived and designed the study and revised the manuscript; Wang YT collected medical records and wrote the manuscript; Zhang Y collected medical records and participated in its design; Tang T collected medical records and provided pictures; Luo C participated in study design and coordination; Liu MY, Xu L,and Wang L collected and organized the literature; All authors read and approved the final manuscript.
All study participants, or their legal guardian, provided informed written consent prior to study enrollment.
The authors declare that they have no conflict of interest.
The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See:https://creativecommons.org/Licenses/by-nc/4.0/
China
Ya-Ting Wang 0000-0003-3498-6181; Yu Zhang 0000-0003-1124-6543; Tao Tang 0000-0002-6467-0922;Chong Luo 0000-0002-7271-3064; Ming-Yue Liu 0000-0002-1934-778X; Li Xu 0000-0002-7288-1607; Li Wang 0000-0003-4663-102X; Xue-Mei Tang 0000-0002-6658-8084.
I looked at her quizzically. What do you mean? Of course you put them out. They were sitting on the porch in a paper bag, right where you said they d be.
Gong ZM
A
Gong ZM
 World Journal of Clinical Cases2022年11期
World Journal of Clinical Cases2022年11期
- World Journal of Clinical Cases的其它文章
- Pleomorphic adenoma of the left lacrimal gland recurred and transformed into myoepithelial carcinoma after multiple operations:A case report
- Thyrotoxicosis after a massive levothyroxine ingestion:A case report
- Contrast-enhanced ultrasound manifestations of synchronous combined hepatocellular-cholangiocarcinoma and hepatocellular carcinoma:A case report
- Papillary thyroid microcarcinoma with contralateral lymphatic skip metastasis and breast cancer:A case report
- Del(5q) and inv(3) in myelodysplastic syndrome:A rare case report
- Ultrasound-guided local ethanol injection for fertility-preserving cervical pregnancy accompanied by fetal heartbeat:Two case reports