Cytochrome P450 family 17 subfamily A member 1 mutation causes severe pseudohermaphroditism:A case report
2022-06-23 02:06:52YuGongFangQinWenJiaLiLeYuLiPingHeXingJianZhou
World Journal of Clinical Cases 2022年11期
lNTRODUCTlON
17 α-Hydroxylase deficiency (17-OHD) is a rare type of congenital adrenal hyperplasia (CAH), which is caused by mutations in the cytochrome P450 family 17 subfamily A member 1 () gene, and the incidence of this disorder is approximately 1 in 50000[1]. In 1966, Biglieri[2] reported the first case of 17-OHD. To date, about 200 cases have been reported at worldwide[3-5]. At present, there is no unified standard for the diagnosis of 17-OHD, which is mainly based on the clinical manifestations, laboratory and imaging examinations,., and the diagnosis depends on the detection of gene[6]. We here report a patient with 17-OHD admitted to our hospital, and review the literature on the pathogenesis, clinical characteristics, diagnosis and treatment of the disease.
CASE PRESENTATlON
Chief complaints
A 29-year-old female was admitted to the Department of Neurology in our hospital due to limb weakness for 1 d.
History of present illness
The patient had a history of syncope on several occasions, which lasted approximately 1 min and could be relieved without treatment.
History of past illness
She denied other medical history such as hypertension or coronary heart disease and had no history of smoking or alcohol consumption.
But the whole shop, from the till down to the shavings, from that night changed their opinion of the tub, and they looked up to it, and had such faith in it that they were under the impression that when the grocer read the art and drama critiques out of the paper in the evenings, it all came from the tub
Personal and family history
Upon further investigation, the patient had primary amenorrhea and was unmarried and childless. Her parents were first cousins and her older brother was healthy.
Physical examination
CAH is an autosomal recessive disorder caused by mutations in the genes encoding essential enzymes for the synthesis of corticosteroids. As a result of the imbalance between glucocorticoids and mineralocorticoids, this leads to metabolic disorders, and thus morbidity and mortality in these patients are very high[9]. The enzymes involved include 21-hydroxylase, 11β-hydroxylase, 17α-hydroxylase, 3βhydroxysteroid dehydrogenase/isomerase. These enzyme defects (reduced or absent activity) can lead to CAH, but with different clinical manifestations[10]. Of these enzymes, 21-hydroxylase deficiency is the most common, accounting for more than 95% of cases[11], followed by 11β-hydroxylase deficiency. 17-OHD accounts for about 1% of all CAH cases, with an estimated incidence of 1 in 50000 to 100000[12,13].
Laboratory examinations
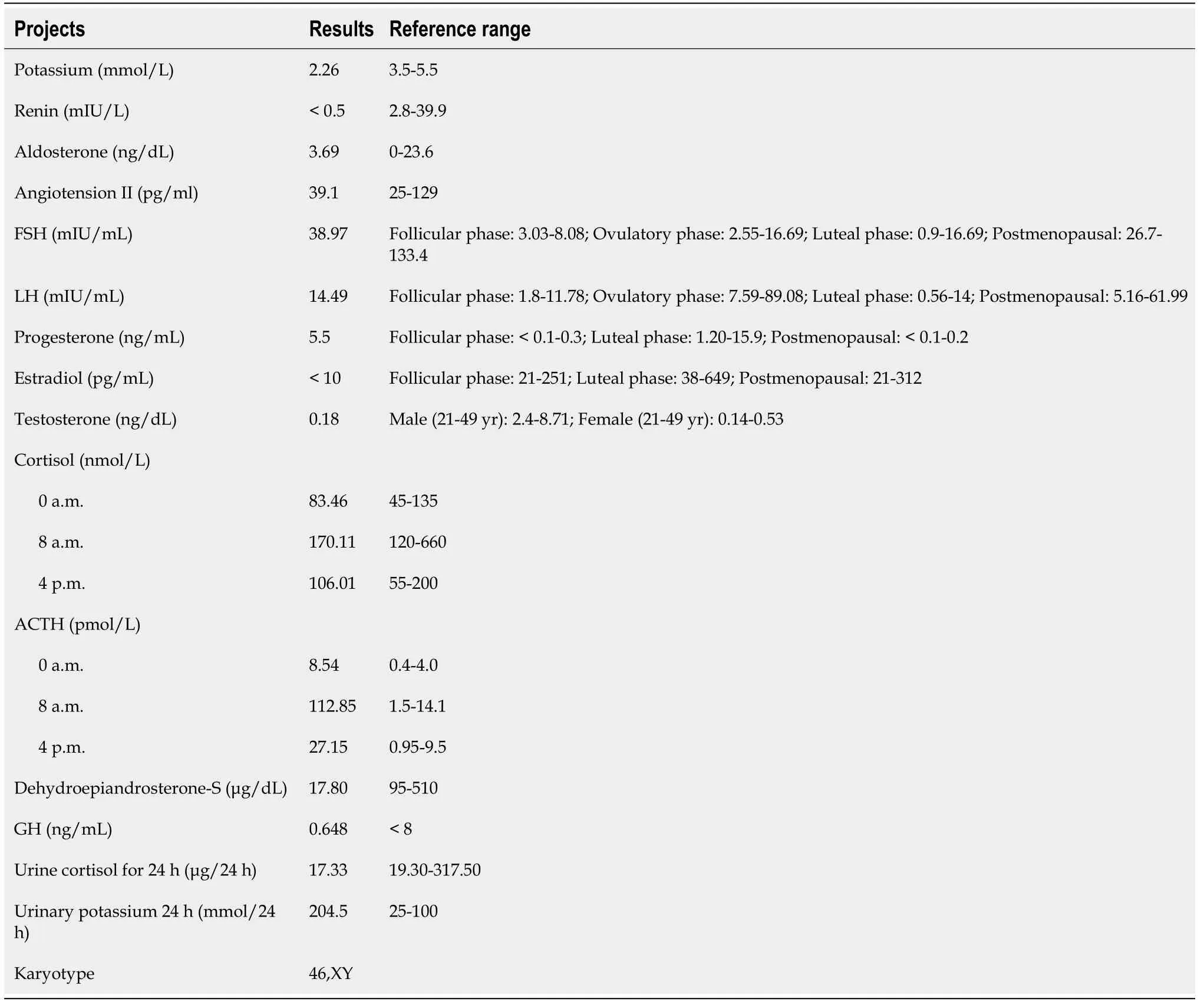
One year later, the patient’s electrolytes (serum potassium level 4.6 mmol/L) and blood pressure(130/75 mmHg) were normal on re-examination.
Imaging examinations
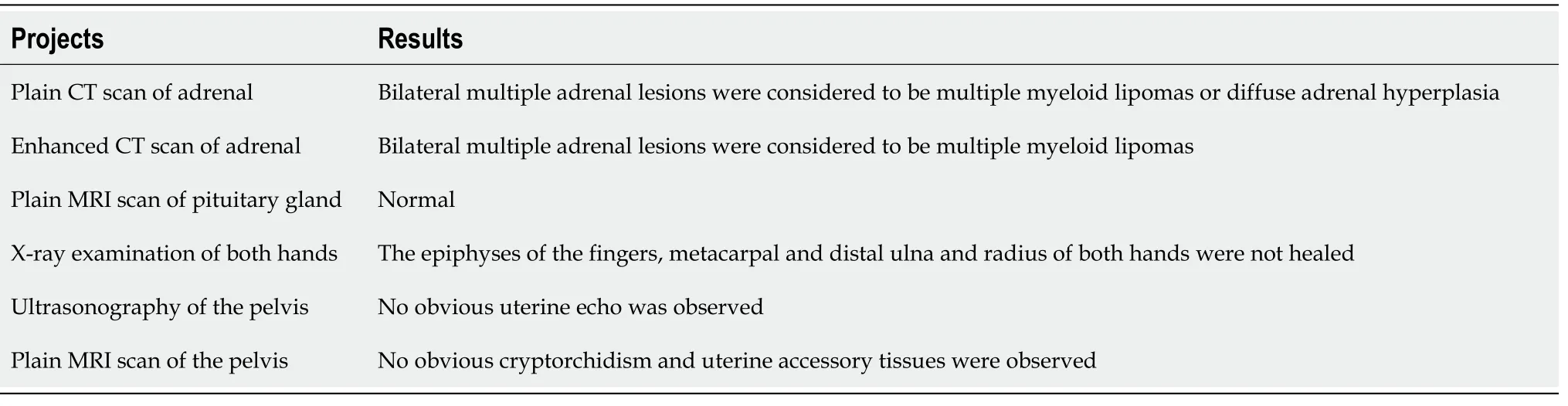
The results of the patient’s biochemical and imaging examinations are shown in Figure 2 and Tables 1 and 2. Genetic analysis (Figure 3) showed homozygous mutations in the CYP17A1 gene (NM_000102.3:c.81C>A (p. Tyr27*)). These genetic variations have been reported by Müssig[7] and Keskin[8].Genetic analysis of the patient showed a heterozygous mutation (c.81C>A). Unfortunately, we were unable to perform a genetic analysis of the patient’s older brother, as he was unavailable at the time of testing.
In summary, 17-OHD is extremely rare in clinical practice, and is prone to misdiagnosis and missed diagnosis. For patients with abnormal development of hypertension and hypokalemia, attention should be paid to the differentiation of this disease. Chromosome karyotype analysis and gene sequencing can help in diagnosis. Hormone replacement and antihypertensive treatment should be given as soon as possible after diagnosis. In addition, the psychological state of patients should be closely monitored.
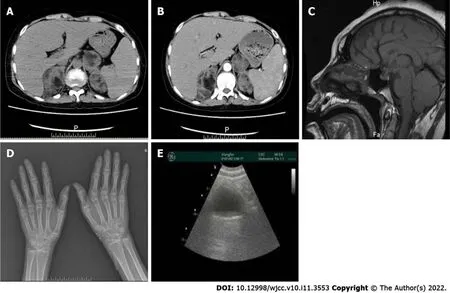

FlNAL DlAGNOSlS
The clinical manifestations of this patient combined with the results of various auxiliary examinations,resulted in a final diagnosis of 17-OHD associated with multiple myeloid lipomas of the adrenal gland.
These days having a best friend seems so important to girls. You want to be special. However I have learned1 the hard way that having one best friend is not the way to go. It s so much better to have many great friends.
TREATMENT
Following the diagnosis of 17-OHD, the patient started on oral dexamethasone (0.75 mg/d), which will be a lifelong medication. When her blood pressure and potassium level had returned to normal, she was discharged from the hospital.
OUTCOME AND FOLLOW-UP
After admission, low serum potassium levels (2.26 mmol/L) were observed and appeared to be uncorrected after potassium supplementation. In addition, the patient had constant high blood pressure,with a maximum reading of 184/127 mmHg.
I discovered that I liked my androgynous() body. It fit my personality - my aggressive male side that loves getting dressed in a helmet, arm guards and shin protectors to do battle with the slalom gates, and my gentle female side that longs to have children one day and wants to dress up in a beautiful silk dress, go out to dinner with a lover and then lie back and be slowly undressed by him.
DlSCUSSlON
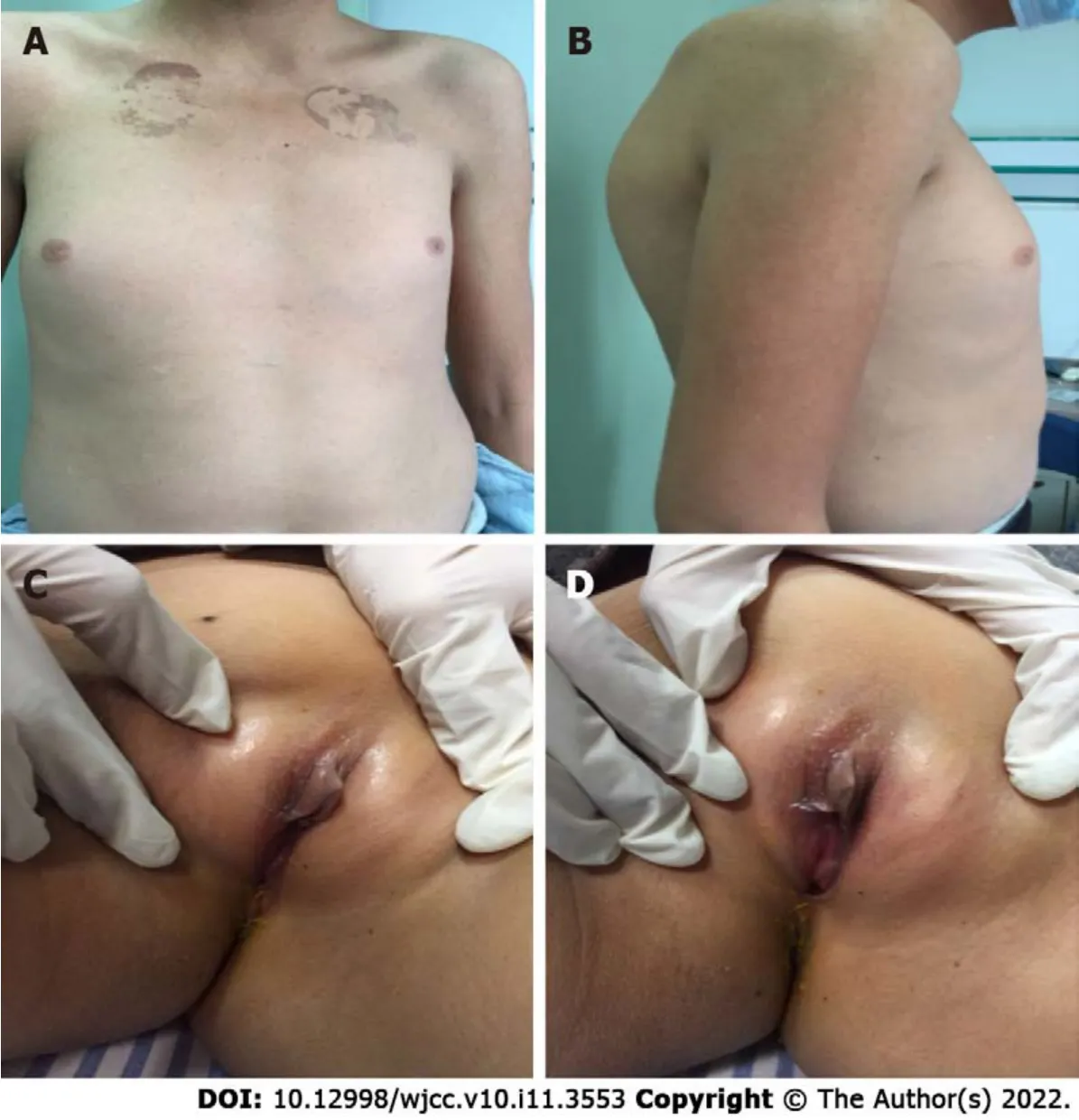
At admission, her temperature was 36.5 °C, respiration rate was 23 breaths/min, pulse rate was 114 bpm, blood pressure was 184/127 mmHg, height was 180 cm, weight was 69 kg, and body mass index was 21.3 kg/m. The patient’s breast development had only progressed to Tanner stage 1, and her vulva was similar to that of a female infant. The patient’s pubic and axillary hair was undeveloped, and her Adam's apple was small. Pathological reflexes were not elicited (Figure 1).
There is no complete cure for 17-OHD, and treatment mainly consists of appropriate glucocorticoid and sex steroid hormone supplementation, social sex selection and psychological interventions. Lowdose glucocorticoid (dexamethasone or prednisolone) replacement therapy is administered in order to decrease and normalize the blood levels of 11-DOC and ACTH, which can normalize blood pressure and electrolyte imbalances[21]. However, due to the need to avoid high-dose glucocorticoid therapy and complete inhibition of the hypothalamic–pituitary–adrenal axis, DOC cannot be completely suppressed,and many patients will eventually become hypertensive, and corticosteroid receptor antagonists or calcium channel blockers can be used to control blood pressure[11]. Therefore, during treatment,spironolactone and nifedipine sustained release tablets are given to control blood pressure. In addition,sex hormone replacement is required for breast and uterus development and to maintain female sexual characteristics. Patients require estrogen and progestin circulation therapy to induce circulatory arrest bleeding and prevent endometrial hyperplasia. If the patient decides to be considered male, androgen replacement therapy may be given, and extensive genital reconstructive surgery may be performed,such as gonadectomy, to avoid malignant degeneration of the testes within the abdomen[13]. In this case, after full communication with the patient and her family, she decided to temporarily discontinue sex hormones and surgical treatment.
17 -OHD mainly manifests as hypertension, hypokalemia and abnormal sexual development.encodes an enzyme with 17a-hydroxylase and 17,20-lyase activities, which is essential for the normal production of adrenal and gonadal glands[14]. When it is deficient, pregnenolone cannot translate into 17-hydroxyprogesterone and 17-hydroxylpregnenolone, resulting in the impairment of cortisol and gonadal hormones (including testosterone and estrogen)[13]. Cortisol synthesis disorders lead to an increase in adrenocorticotropic hormone (ACTH) feedback, which further activates the 17-deoxy pathway of the zona fasciculata, producing overstimulation of this pathway and increasing progesterone, corticosterone and deoxycorticosterone (DOC) synthesis. The excessive levels of these hormones then lead to hypertension, and hypokalemia. Deficiency of gonadal hormones causes primary amenorrhea in women[15] and feminization of external genitalia in men[16].Sexual dysplasia[6] in male patients mostly manifests as pseudohermaphroditism, with infantile female genitalia and a blind end vagina, while the internal genitalia is of the male type with small testicles and dysplasia, and external genitals are difficult to distinguish, such as small penis or mammary gland development. Female patients can be normal at birth, but do not develop secondary sexual signs with primary amenorrhea.There is no pubic or axillary hair growth in both men and women. After puberty, both folliclestimulating hormone and luteinizing hormone are significantly increased. Due to the lag in bone age,the patient's height continues to increase slowly after reaching adulthood. The bone age of our patient was below the actual age, but she was tall (180.0 cm). In addition, some patients are prone to fatigue,infection, different degrees of skin pigmentation[17] and osteoporosis. The diversity of clinical manifestations in 17-OHD patients is due to the different mutation sites on the gene encoding the enzyme and different effects on the enzyme function. Therefore, in the clinic, the existence of hypertension, and hypokalemia accompanied by sexual dysplasia, should be considered as possible 17-OHD. This deficiency should be distinguished from several other diseases, such as 5α-reductase deficiency,androgen insensitivity syndrome and 3β-hydroxysteroid dehydrogenase deficiency. However, both 5αreductase deficiency and androgen insensitivity syndrome are generally not accompanied by hypertension and hypokalemia[18,19], and the clinical manifestations in this case did not meet the criteria for 3 β-hydroxysteroid dehydrogenase deficiency[20]. Hence, we were able to rule out these diseases.
CONCLUSlON
Of course, at the very mention of the Enchanter as a rival he was furious, and I don t know what foolish things he would not have done if Melinette had not been there to calm him down
Gong Y and Qin F treated the patient; Zhou XJ drafted the manuscript; Li WJ and Li LY participated in the analysis and the interpretation of the data; He P critically revised the manuscript; all authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
When I married, I told my wife Susan about the significant part the lowly pickle jar had played in my life as a boy. In my mind, it defined, more than anything else, how much my dad had loved me. No matter how rough things got at home, Dad continued to doggedly15 drop his coins into the jar. Even the summer when Dad got laid off from the mill, and Mama had to serve dried beans several times a week, not a single dime10 was taken from the jar. To the contrary, as Dad looked across the table at me, pouring catsup over my beans to make them more palatable16, he became more determined17 than ever to make a way out for me. When you finish college, son, he told me, his eyes glistening18, you ll never have to eat beans again unless you want to.
The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
The authors declare that they have no conflict of interest.
Informed written consent was obtained from the patient for publication of this report and any accompanying images.
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See:https://creativecommons.org/Licenses/by-nc/4.0/
Therefore it was with great joy he heard that she was living and that a king s son asked her in marriage, and he quitted his kingdom with his elder daughter so as to be present at the ceremony
China
Yu Gong 0000-0001-7246-0637; Fang Qin 0000-0003-1173-2592; Wen-Jia Li 0000-0001-9609-968X; Le-Yu Li 0000-0002-4500-932X; Ping He 0000-0002-8060-6840; Xing-Jian Zhou 0000-0001-9432-111X.
Gao CC
A
The accomplices21 willingly accepted this invitation; and after Simon had made all his purchases, he tied them on to the goat s back, and said to it, in the presence of the three cheats, Go home now, and tell Nina to roast the veal, and boil the chickens, and tell her to prepare a savoury with herbs, and to bake the best tart she can make
Gao CC
 World Journal of Clinical Cases2022年11期
World Journal of Clinical Cases2022年11期
- World Journal of Clinical Cases的其它文章
- Pleomorphic adenoma of the left lacrimal gland recurred and transformed into myoepithelial carcinoma after multiple operations:A case report
- Thyrotoxicosis after a massive levothyroxine ingestion:A case report
- Contrast-enhanced ultrasound manifestations of synchronous combined hepatocellular-cholangiocarcinoma and hepatocellular carcinoma:A case report
- Papillary thyroid microcarcinoma with contralateral lymphatic skip metastasis and breast cancer:A case report
- Del(5q) and inv(3) in myelodysplastic syndrome:A rare case report
- Ultrasound-guided local ethanol injection for fertility-preserving cervical pregnancy accompanied by fetal heartbeat:Two case reports