ABCC8基因復合雜合突變致兒童KATP-HI 9例臨床分析
2022-06-17 07:48:40曾俏徐子迪張琳吳玉筠劉敏閆潔桑艷梅朱逞倪桂臣
疑難病雜志 2022年6期
曾俏,徐子迪,張琳,吳玉筠,劉敏,閆潔,桑艷梅,朱逞,倪桂臣
先天性高胰島素血癥(congenital hyperinsulinism,CHI)是新生兒和兒童持續復發性低血糖常見的原因之一,是一種因胰島素分泌調節異常引起的遺傳異質性疾病。隨著研究的不斷進展,迄今已發現了15種CHI相關的致病基因,主要包括ABCC8、KCNJ11、GLUD1等,相應地構成14種遺傳學類型。其中,ATP敏感鉀通道型先天性高胰島素血癥(adenosine triphosphate-sensitive potassium channel hyperinsulinism,KATP-HI)是CHI最常見的類型,是因ABCC8和KCNJ11基因失活突變導致[1],這2個基因分別編碼磺脲類受體1蛋白(sulfonylurea receptor1,SUR1)和內向整流鉀通道蛋白(potassium inward rectifying channel,Kir6.2)。迄今為止,ABCC8基因已發現了400多種突變,KCNJ11基因已發現了60多種突變[2]。其中,ABCC8基因復合雜合突變臨床較為少見,迄今國內外報道的只有幾十例。導致KATP-HI的KCNJ11復合雜合突變臨床報道目前僅3例[3-5]。本研究搜集攜帶ABCC8復合雜合突變的9例KATP-HI 患兒為研究對象,對其臨床特征及遺傳學特征進行分析,以期提高臨床醫師對該病的認識。
1 臨床資料
1.1 一般資料 選取2012年5月—2017年8月北京兒童醫院內分泌遺傳代謝科收治的攜帶ABCC8基因復合雜合突變KATP-HI患兒9例,均符合CHI的診斷標準。具體診斷標準如下[6]:(1)高胰島素血癥(血漿胰島素>2 mIU/L);(2)低脂肪酸血癥(血漿游離脂肪酸 <1.5 mmol/L);(3)低酮血癥(血漿β-羥丁酸 <2.0 mmol/L);(4)1 mg靜脈胰高血糖素試驗反應:血糖變化>1.7 mmol/L。其中男6例,女3例。起病年齡生后1 d~2歲,中位起病年齡為生后1 d。出生體質量1.8~4.9 kg,正常出生體質量兒2例,巨大兒6例,低出生體質量兒1例,見表1。
1.2 臨床特征 首發癥狀為無明顯誘因抽搐3例,有低血糖癥狀3例(如反應弱、頭面部青紫和四肢肌張力減低等),無癥狀性低血糖3例。1例患兒(例9)的姐姐患有CHI,于5歲時起病,與患兒攜帶相同突變。其余8例患兒無低血糖家族史。9例均無糖尿病家族史及母孕期糖耐量受損史。2例(例1和例4)曾分別于香港養和醫院和復旦大學附屬華山醫院進行18-氟左旋多巴正電子發射斷層(18F-L-DOPA PET/CT)胰腺掃描,胰腺組織學類型均提示彌漫型。
1.3 基因突變攜帶情況 9例患兒均攜帶ABCC8基因(NM_000352)復合雜合突變,見表1。例2的p.R1245G、p.N72K,例3的p.A471V、p.L1211P,例4的p.T1042QfsTer75、p.E945RfsTer25,例7的p.A640V、p.Q1196*及例9的p.P1161R突變為中國人首次報道。
1.4 治療與轉歸 二氮嗪治療8例,有效3例,無效5例。2例在應用二氮嗪治療期間出現血小板減少、中性粒細胞絕對值減少、低血鉀、納差等不良反應。奧曲肽治療4例,均有效,治療過程中1例患兒出現肝功能損害。行胰腺次全切除術治療者1例(例4)。經長期隨訪,3例通過少量多餐飲食維持血糖,空腹時間延長時,偶有低血糖發作;2例長期奧曲肽治療,3例自行緩解,1例失訪,見表1。
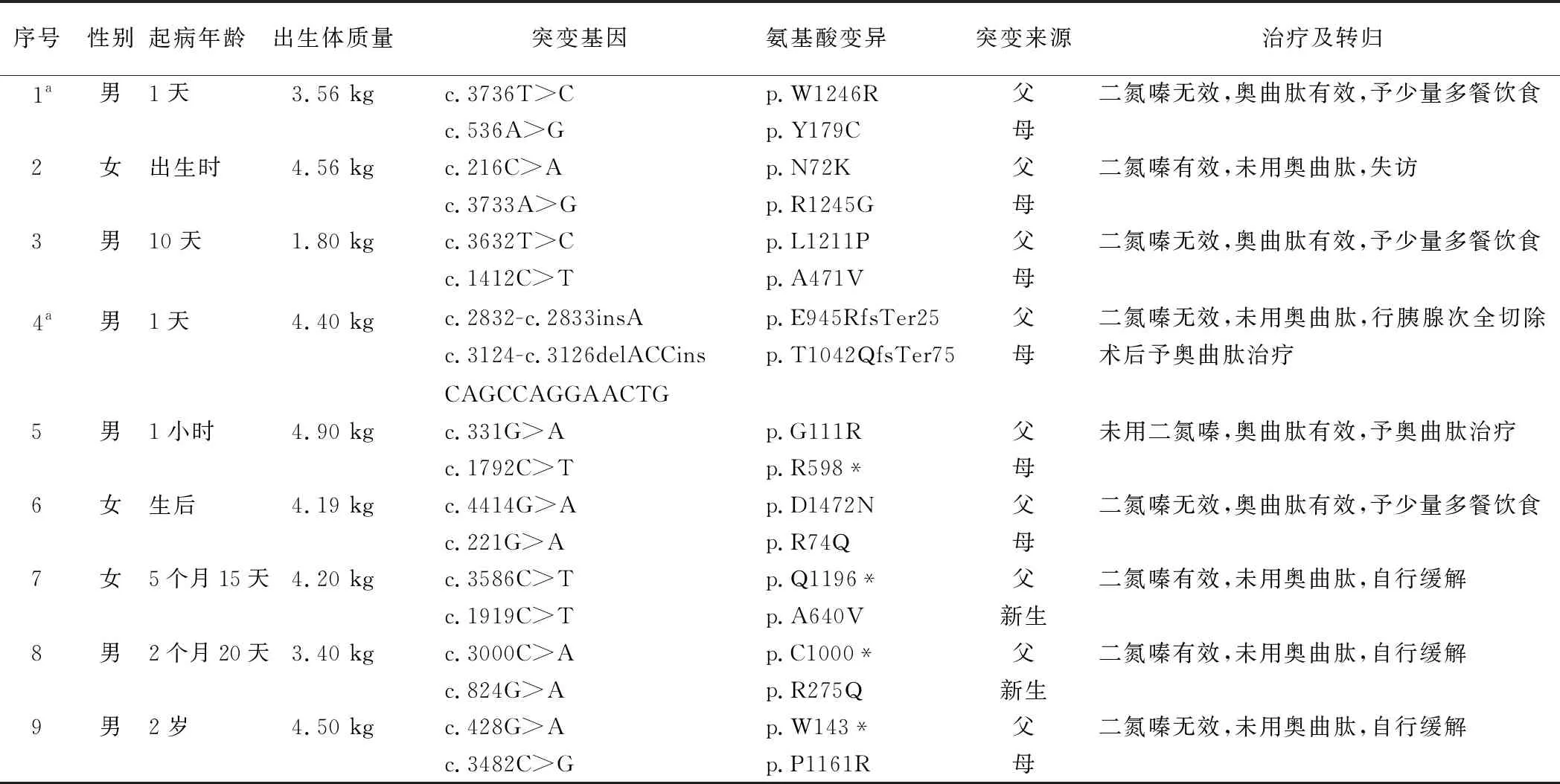
表1 9例KATP-HI患兒攜帶ABCC8基因復合雜合突變情況及主要臨床特征
例4于1歲11個月時行胰腺次全切除術(90%),術后仍有低血糖發作,故予奧曲肽長期治療維持正常血糖水平,目前4歲,奧曲肽劑量5 μg·kg-1·d-1。例9于5歲時自行緩解,隨訪過程中無低血糖發作,自2歲起診斷癲癇,服用抗癲癇藥物至今,仍有間斷抽搐發作,頻率4~5次/年,每次持續約 2 min緩解。例3于3歲時自行停用奧曲肽,空腹時間延長時偶有低血糖及抽搐發作,發作頻率約每2個月1次,口服葡萄糖水后1~2 min緩解,現4歲,語言發育遲緩。例5出院后奧曲肽治療至今,納差時出現低血糖發作(1.9~2.4 mmol/L),現3歲,語言發育遲緩。
2 討 論
KATP-HI是CHI最常見和最嚴重的類型,其遺傳方式多為常染色體隱性遺傳,少數為常染色體顯性遺傳,還有一部分為新生突變。
ABCC8和/或KCNJ11基因復合雜合突變導致的KATP-HI臨床較罕見,迄今只報道了60多例。其中,多數(93%)為ABCC8基因復合雜合突變,少數(4%)為KCNJ11基因復合雜合突變。另外,還報道了2例(3%)ABCC8/KCNJ11復合雜合突變。本研究中,9例患兒均攜帶ABCC8基因復合雜合突變,經遺傳學確診為KATP-HI,符合文獻報道的復合雜合突變基因分布特征。
隨著研究不斷深入,迄今已報道52種ABCC8復合雜合突變,較常見的突變包括p.A640V、p.R168C、p.Q1196*、p.R598*等。報道過的復合雜合突變中,只有1例為新生突變(c.IVS10+1G>T/ p.T1531A),其中p.T1531A為新生突變,其余突變多來自表型正常的父母。已報道的ABCC8基因復合雜合突變類型復雜多樣,有錯義突變、移碼突變、剪切突變和缺失突變等。
本組9例患兒共檢測到18個ABCC8基因突變,遺傳方式均為常染色體隱性遺傳。其中9個突變已有文獻報道,另9個為中國人首次報道,尚未見相關數據庫收錄(參考數據庫:Pubmed及ClinVar數據庫),其變異均可能有致病性。除p.A640V、p.R275Q突變為新生突變外,其余16個突變均來自臨床表型正常的父母,符合文獻報道的ABCC8基因復合雜合突變特征。本組病例突變類型共3種,錯義突變:p.Y179C、p.W1246R、p.N72K等12個;無義突變:p.R598*、p.Q1196*等4個;移碼突變:p.T1042QfsTer75、p.E945RfsTer25。
文獻資料顯示,攜帶ABCC8基因復合雜合突變的CHI患者多起病較早,多于新生兒期起病,少數于嬰兒期起病。迄今報道資料完善的42例患者中[3-5,7-18],新生兒期起病38例(90.5%),1~6個月起病3例(7.1%),6個月后起病1例(2.4%)。本組患兒中,新生兒期起病6例(66.7%),1~6個月起病2例(22.2%),6個月后起病1 例(11.1%),基本符合文獻報道的起病年齡特征。
攜帶ABCC8基因復合雜合突變的KATP-HI患兒出生體質量多為巨大兒[3-5,7-18]。在既往報道出生體質量明確的33例病例中,低出生體質量兒1例(3.0%),正常出生體質量兒8例(24.2%),巨大兒24例(72.7%)。本研究中,低出生體質量兒1例(11.1%),正常出生體質量兒2例(22.2%),巨大兒6例(66.7%),出生體質量占比與文獻報道基本一致。
根據胰腺組織學特征的不同,KATP-HI可分為彌漫型、局灶型和非典型3種類型。既往研究顯示,攜帶ABCC8基因復合雜合突變的CHI患兒組織分型大多數為彌漫型(89%),少數為局灶型(7%),只有1例(4%)為非典型[3-5,7-18]。Zhang等[7]于2015年首次報道1例中國漢族人ABCC8基因復合雜合突變病例,該例術前18F-L-DOPA-PET胰腺掃描顯示胰頭有局灶性病變,胰腺病理提示為非典型,這也是目前唯一1例非典型的ABCC8基因復合雜合突變病例。本組僅2例行18F-L-DOPA-PET胰腺掃描,結果均為彌漫型。其余7例組織學分型尚不明確,根據既往研究資料推測為彌漫型可能性大。
KATP-HI治療分為內科治療和外科治療。二氮嗪是CHI治療的一線治療藥物,二氮嗪無效患兒可選用奧曲肽[8]。大多數ABCC8和/或KCNJ11導致的KATP-HI對二氮嗪藥物治療無效,需行不同程度的胰腺切除術[9]。功能缺陷的KATP通道導致質膜持續去極化,胰島素大量分泌,二氮嗪作為一種KATP通道激動劑,使通道處于開放狀態,從而抑制胰島素分泌,部分突變的復合雜合子之間相互作用,導致細胞內保留通道復合物,故二氮嗪仍能發揮作用[19]。
本研究對既往報道ABCC8基因復合雜合突變的文獻進行統計[3-5,7-18],大多數ABCC8基因復合雜合突變導致的CHI對二氮嗪藥物治療無效,27例中有效占15%,無效占85%。本研究中,8例患兒曾應用二氮嗪試驗性治療,有效3例(37.5%),無效5例(62.5%),與文獻報道基本一致。文獻資料顯示,ABCC8基因復合雜合突變導致的7例CHI患兒,應用奧曲肽治療部分有效,有效占43%,無效占57%。本組4例曾應用奧曲肽治療,均有效,這種有效性的差異,考慮與本研究的樣本量較小有關。既往報道的60多例ABCC8基因復合雜合突變病例中[3-5,7-18],23例曾行手術治療,僅15例描述了術后情況,其中,術后血糖穩定者2例(13%),術后并發糖尿病需胰島素治療者3例(20%),術后仍有反復低血糖發作者10例(67%)。本組病例中,1例經胰腺次全切除術治療,術后仍有低血糖發作,需長期奧曲肽治療以維持正常血糖水平。
研究資料顯示,ABCC8基因復合雜合突變患兒大多數預后不良,可能出現發育遲緩、語言障礙、癲癇、智力低下等,早產兒持續反復的低血糖發作與18個月大時發生腦癱、發育遲緩、智力低下的風險增高有關,部分患兒經長期藥物治療及飲食控制,血糖維持平穩[13,20]。本研究中,例9于2歲時起病,以抽搐為首發癥狀,隨后診斷為癲癇,提示低血糖對中樞神經系統的損傷可能在疾病診斷之前就已經發生。例3和例5有語言發育遲緩,其中例3為36+5周早產兒,為低出生體質量兒,生后10 d以抽搐和低血糖癥狀起病,目前納食少或禁食6 h情況下出現抽搐發作,語言發育遲緩可能是因為患兒為早產兒,且喂養不當,導致嚴重反復低血糖發作對神經系統造成不可逆損傷。
部分ABCC8基因復合雜合突變患兒通過保守治療低血糖癥狀可得到改善,且預后較好,甚至可自行緩解[10]。本研究中,3例(例7、8、9)自行緩解,自行緩解率為33.3%,其中例7和例8對二氮嗪敏感,分別于1歲和 2歲時自行緩解。例9二氮嗪治療無效,5歲時低血糖癥狀自行緩解,但合并癲癇,需長期口服抗癲癇藥物治療。
總之,本研究資料顯示,中國KATP-HI兒童的ABCC8基因復合雜合突變類型多樣,以錯義突變為主。攜帶ABCC8基因復合雜合突變的患兒多為巨大兒,多于新生兒期發病。該型患兒多對二氮嗪治療不敏感,部分對奧曲肽治療有效,部分患兒可獲得自行緩解。
猜你喜歡
人人健康(2023年26期)2023-12-07 03:55:46
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
中國生殖健康(2019年2期)2019-08-23 08:12:10
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
