HPLC法同時測定小兒七星茶顆粒中9種鉤藤成分
2022-06-15 07:26:54陳素云趙曉彤曹依敏黃曉靜
中成藥 2022年5期
陳素云, 周 恒, 趙曉彤, 曹依敏, 黃曉靜, 季 申*
(1.上海中醫藥大學中藥學院,上海 201203;2.上海市食品藥品檢驗研究院,國家藥品監督管理局中藥質量控制重點實驗室,上海 201203)
小兒七星茶顆粒是常用小兒消食藥,由薏苡仁、山楂、稻芽、淡竹葉、鉤藤、蟬蛻、甘草7味中藥組成,具有開胃消滯、清熱定驚的功效,臨床上主要用于小兒積滯化熱、消化不良、夜寐不安等癥狀的治療[1],收載于2020年版《中國藥典》一部,但現行標準僅對方中甘草、山楂藥味規定了薄層鑒別、含量測定等項目[2]。課題組前期發現,小兒七星茶顆粒中歸屬于鉤藤的主要色譜峰數量與甘草相當,遠多于其他藥味,但目前缺乏對其所含成分的專屬質量控制方法。因此,本實驗在前期研究的基礎上結合文獻[3-5],建立HPLC法同時測定小兒七星茶顆粒中喜果苷、二氫卡丹賓、異去氫鉤藤堿、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚9種鉤藤成分的含量,以期為該制劑的質量控制提供依據。
1 材料
Agilent 1260高效液相色譜儀(美國Agilent公司);XSE105電子分析天平(瑞士Mettler-Toledo公司);Elmasonic E100H超聲波清洗儀(德國Elma公司);Milli-Q型超純水系統(美國Milipore公司)。喜果苷對照品(上海同田生物技術股份有限公司,批號20081424,純度99.4%);二氫卡丹賓(批號6072,純度98.2%)、異去氫鉤藤堿(批號2557,純度99.9%)、去氫鉤藤堿(批號4075,純度98.9%)、去氫毛鉤藤堿(批號4170,純度98.9%)、毛鉤藤堿(批號6068,純度98.9%)、縫子嗪甲醚(批號6069,純度98.1%)對照品(上海詩丹德生物技術有限公司);異鉤藤堿(批號111927-201102)、鉤藤堿(批號112028-201601)對照品(中國食品藥品檢定研究院)。小兒七星茶顆粒共56批(編號1~56),均為市售,具體信息見表1,缺鉤藤陰性樣品由相關企業提供。乙腈、甲醇為色譜純(德國Merck公司);二乙胺、氨水為分析純(上海凌峰化學試劑有限公司)。
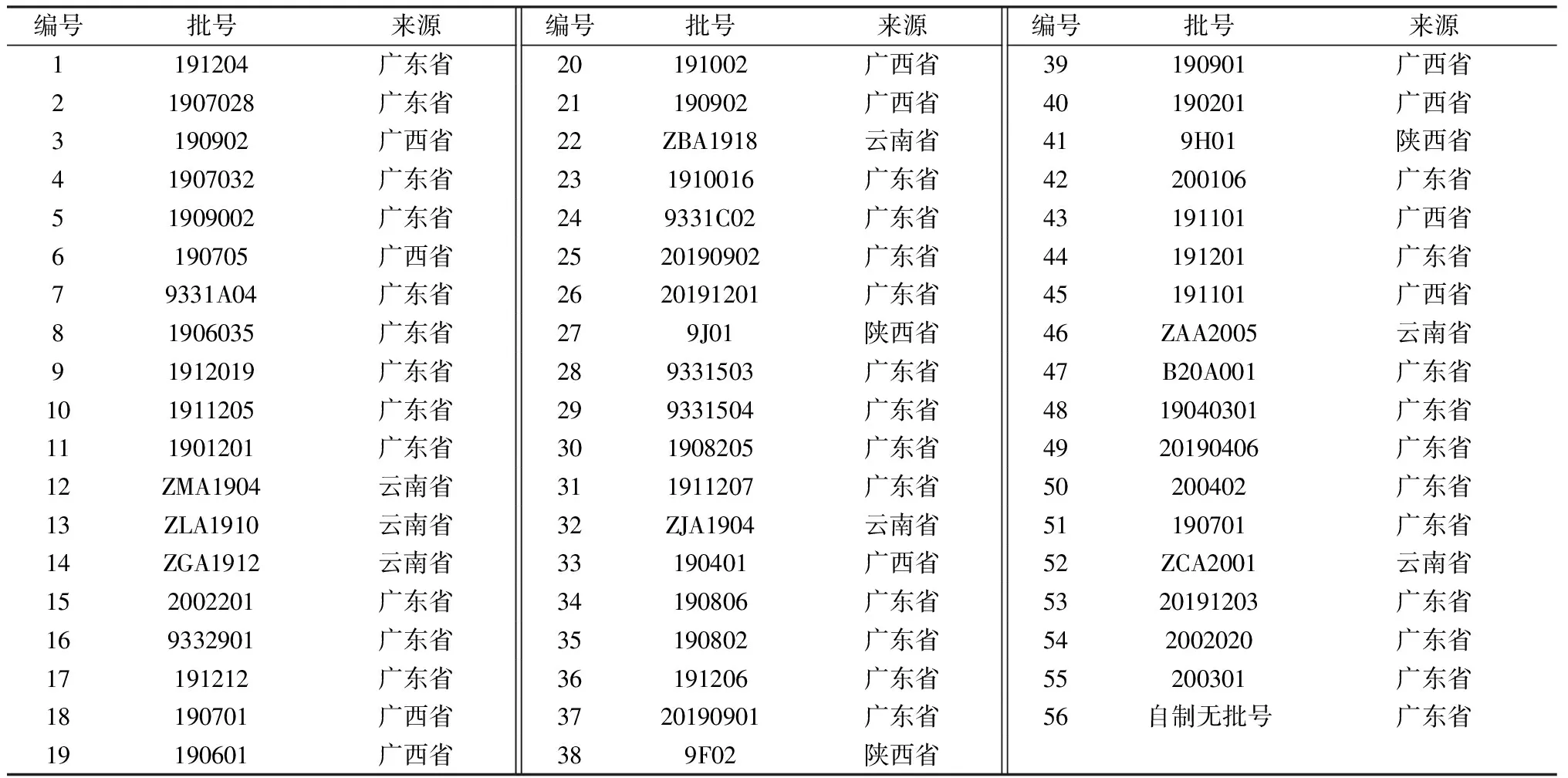
表1 樣品信息
2 方法與結果
2.1 色譜條件 Agilent Extend C18色譜柱(4.6 mm×250 mm,5 μm);流動相乙腈(A)-0.015%二乙胺(B),梯度洗脫,程序見表2;體積流量1.0 mL/min;柱溫30 ℃;檢測波長246 nm;進樣量10 μL。
2.2 溶液制備
2.2.1 對照品溶液 精密稱取喜果苷、二氫卡丹賓、異去氫鉤藤堿、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚對照品適量,加乙腈制成每1 mL各含1 mg上述成分的貯備液,精密量取適量,甲醇定容至20 mL,即得(質量濃度為50 μg/mL),并用甲醇逐級稀釋至1、2、5、10、20 μg/mL,在4 ℃下保存。
2.2.2 供試品溶液 取本品粉末約5 g,精密稱定,置于具塞錐形瓶中,精密加入50 mL甲醇,稱定質量,超聲(功率300 W、頻率40 kHz)處理30 min,放冷,甲醇補足減失的質量,搖勻,濾過,精密量取續濾液20 mL,水浴蒸至近干,殘渣加甲醇溶解并定容至2 mL,搖勻,微孔濾膜過濾,取續濾液,即得。
2.2.3 陰性樣品溶液 按工藝及處方制備缺鉤藤的陰性樣品,按“2.2.2”項下方法制備,即得。
2.3 方法學考察
2.3.1 專屬性考察 精密吸取對照品、供試品、陰性樣品溶液各10 μL,在“2.1”項色譜條件下進樣測定,結果見圖1。由此可知,陰性無干擾,表明該方法專屬性良好。
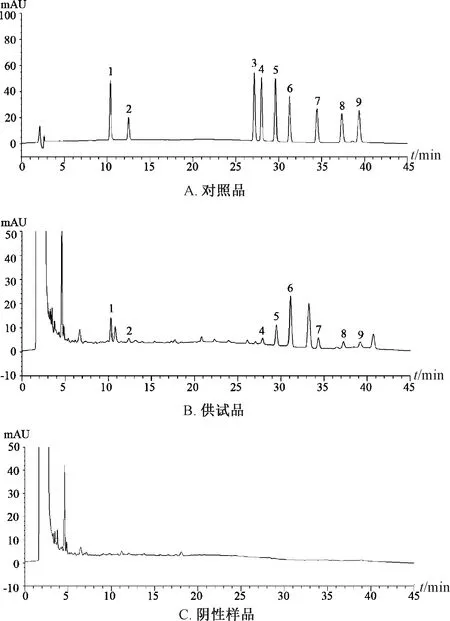
1.喜果苷 2.二氫卡丹賓 3.異去氫鉤藤堿 4.去氫鉤藤堿 5.異鉤藤堿 6.鉤藤堿 7.去氫毛鉤藤堿 8.毛鉤藤堿 9.縫子嗪甲醚1.vincosamide 2.3α-dihydrocadambine 3.isocorynoxeine4.corynoxeine 5.isorhynchophylline 6.rhynchophylline7.corynantheine 8.hirsutine 9.geissoschizine methyl ether圖1 各成分HPLC色譜圖Fig.1 HPLC chromatograms of various constituents
2.3.2 線性關系考察 精密吸取“2.2.1”項下不同質量濃度對照品溶液各10 μL,在“2.1”項色譜條件下進樣測定。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,結果見表3,可知各成分在各自范圍內線性關系良好。
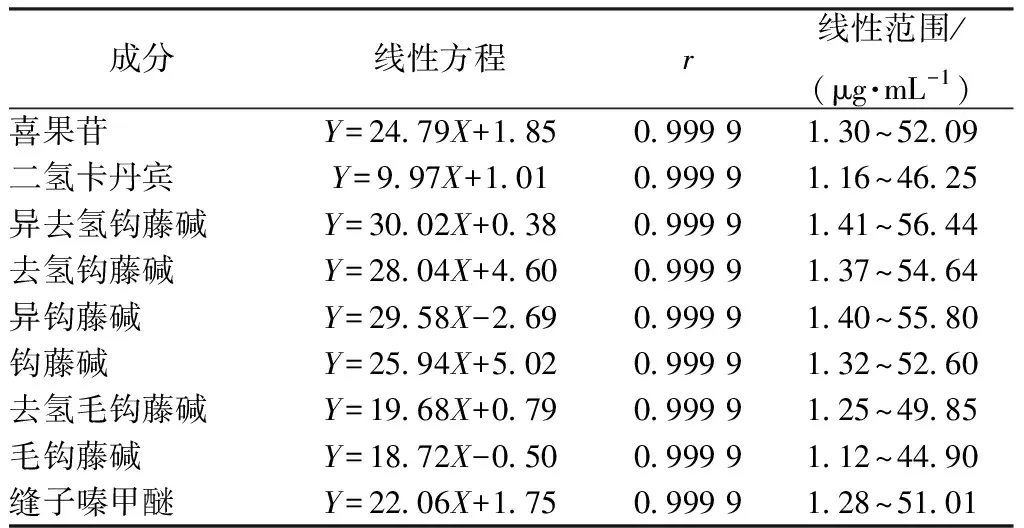
表3 各成分線性關系考察
2.3.3 精密度試驗 精密吸取10 μg/mL對照品溶液10 μL,在“2.1”項色譜條件下進樣測定6次,測得喜果苷、二氫卡丹賓、異去氫鉤藤堿、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚峰面積RSD分別為0.2%、0.3%、0.4%、0.1%、0.7%、0.4%、0.2%、0.7%、0.2%,表明儀器精密度良好。
2.3.4 穩定性試驗
2.3.4.1 對照品溶液 精密吸取10 μg/mL對照品溶液10 μL,于0、1、2、3、4、24 h在“2.1”項色譜條件下進樣測定,測得喜果苷、二氫卡丹賓、異去氫鉤藤堿、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚的峰面積RSD分別為0.4%、0.4%、1.3%、0.8%、2.9%、2.5%、0.3%、0.6%、0.3%,表明溶液在24 h內穩定性良好。
2.3.4.2 供試品溶液 取本品粉末(編號4)適量,按“2.2.2”項下方法制備供試品溶液,精密吸取10 μL,于0、1、3、15、24 h在“2.1”項色譜條件下進樣測定,除異去氫鉤藤堿未檢出外,測得喜果苷、二氫卡丹賓、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚峰面積RSD分別為1.2%、1.5%、1.2%、2.1%、0.2%、0.4%、0.8%、0.8%,表明溶液在24 h內穩定性良好。
2.3.5 重復性試驗 取本品粉末(編號4)適量,按“2.2.2”項下方法平行制備6份供試品溶液,在“2.1”項色譜條件下進樣測定,除異去氫鉤藤堿未檢出外,測得喜果苷、二氫卡丹賓、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚含量RSD分別為2.1%、2.5%、3.7%、1.3%、1.5%、1.4%、1.3%、1.3%,表明該方法重復性良好。
2.3.6 加樣回收率試驗 精密稱取本品粉末(編號4)2.5 g,共9份,置于具塞錐形瓶中,以3份為一組,每組分別精密加入50 μg/mL對照品溶液0.25、0.5、1 mL,按“2.2.2”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣測定,計算回收率。結果,喜果苷、二氫卡丹賓、異去氫鉤藤堿、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚平均加樣回收率分別為95.3%、92.6%、97.4%、98.6%、91.6%、104.9%、97.3%、94.9%、95.7%,RSD分別為5.2%、4.0%、1.8%、5.5%、2.5%、2.8%、2.7%、4.0%、3.8%。
2.4 樣品含量測定 取56批樣品,按“2.2.2”項下方法各平行制備2份供試品溶液,在“2.1”項色譜條件下進樣測定,計算含量,結果見表4。
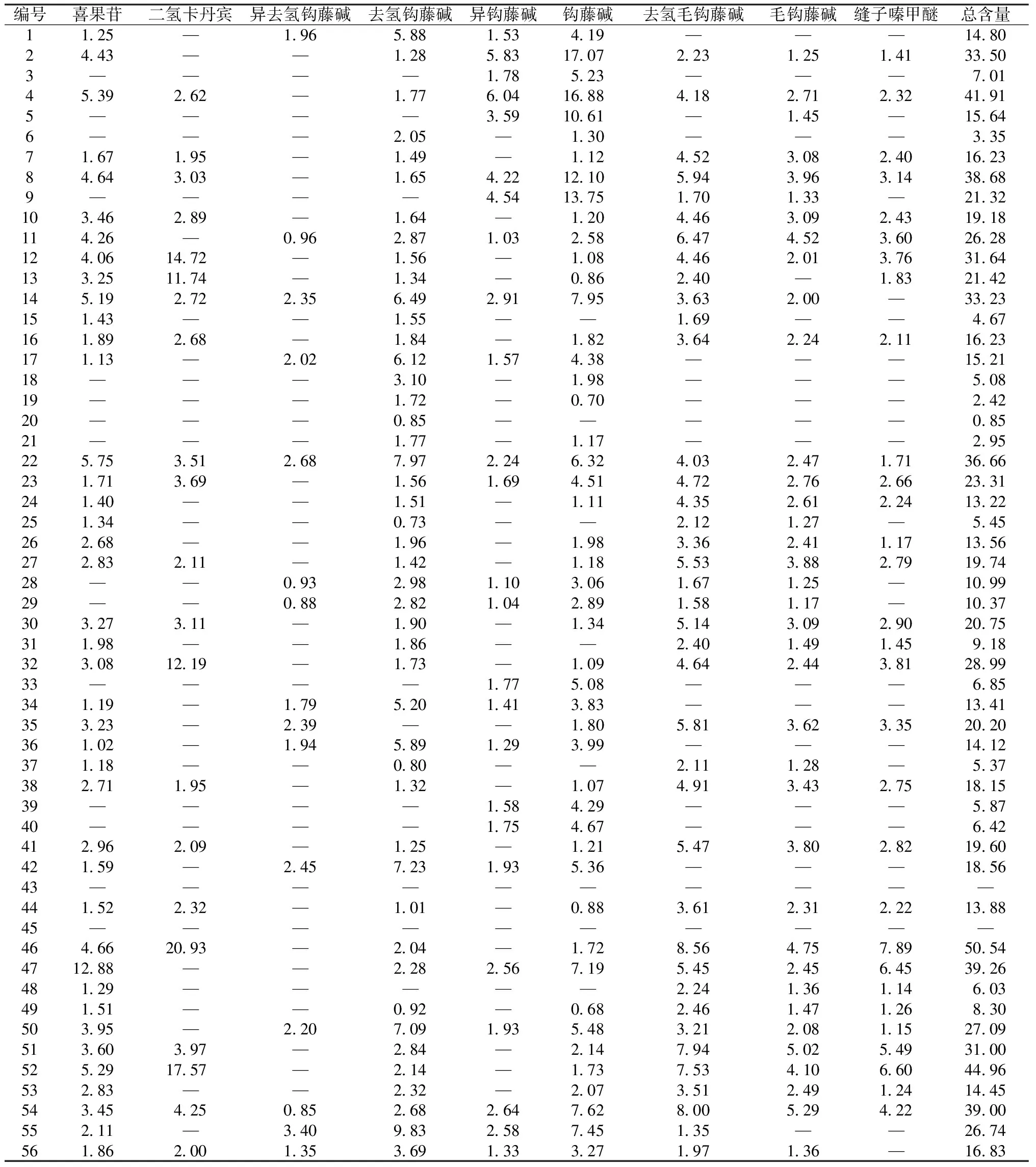
表4 各成分含量測定結果(μg/g,n=2)
3 討論
3.1 色譜條件選擇 本實驗考察了Extend C18(4.6 mm×250 mm,5 μm)、XBridge C18(4.6 mm×100 mm,3.5 μm)、XBridge Shield RP 18(4.6 mm×250 mm,5 μm)、TechMate C18ST(4.6 mm×250 mm,5 μm)色譜柱,發現Extend C18柱對9種鉤藤成分的分離效果最優。再比較了乙腈-0.015%二乙胺[4]、甲醇-0.1%三乙胺(冰醋酸調pH值7.5)[6-8],最終確定前者作為流動相。
3.2 對照品稀釋溶劑選擇 鉤藤中部分生物堿的穩定性較差,對溶劑較敏感,容易發生互變異構[9-12]。本實驗考察了甲醇、70%甲醇對9種鉤藤成分穩定性的影響,最終確定前者作為對照品稀釋溶劑。
3.3 供試品提取溶劑選擇 本實驗考察了甲醇、1%氨水甲醇、氨水-二氯甲烷-甲醇(1∶5∶5)[13]、氨試液-三氯甲烷(2∶7)[14]的提取效果,最終確定甲醇作為供試品提取溶劑。
3.4 含量分析 11個廠家56批樣品中9種鉤藤成分的種類和含量存在較大差異,而且有2批未檢出所有成分,可能與藥材質量、生產工藝等因素有關,值得關注。
4 結論
本實驗首次建立HPLC法同時測定小兒七星茶顆粒中喜果苷、二氫卡丹賓、異去氫鉤藤堿、去氫鉤藤堿、異鉤藤堿、鉤藤堿、去氫毛鉤藤堿、毛鉤藤堿、縫子嗪甲醚9種鉤藤成分的含量,該方法操作簡便,準確度高,重復性好,可為該制劑的質量控制及評價提供科學依據。
