基于GBS-seq的青藏扁蓿豆SNP標記開發
2022-06-09 08:48:14周曉楠徐金青雷雨晴王海慶
生物技術通報 2022年4期
關鍵詞:分析
周曉楠 徐金青 雷雨晴 王海慶
(1.中國科學院西北高原生物研究所 中國科學院高原生物適應與進化重點實驗室,西寧810001;2.中國科學院大學,北京100049;3.青海省作物分子育種重點實驗室,西寧810001)
青藏扁蓿豆(Medicago archiducis-nicolai)是豆科(Fabaceae)苜蓿屬(Medicago)多年生異花授粉二倍體植物[1],分布于青藏高原及其毗鄰的高海拔高寒地區,對寒冷、干旱等極端環境具有極強的適應性[2-3],是青藏高原天然高寒草地上具有馴化潛力的野生牧草種質資源。遺傳多樣性是種內個體間或一個群體內不同個體遺傳變異的總和,研究物種的遺傳多樣性不僅可揭示該物種群體間的遺傳結構、進化關系,以及與環境及地理分布之間的相關性[4],也可以了解物種的遺傳背景及親緣關系,對種質資源的評價利用具有重要意義。
目前,核基因組(ITS[3])、葉綠體基因組(psbA-trnH[3]、trnL-trnF[5])標記以及簡單重復序列(EST-SSR[6-7])等分子標記技術已被應用于青藏扁蓿豆群體遺傳多樣性研究。利用ITS和psbA-trnH遺傳標記對不同海拔的青藏扁蓿豆野生群體進行遺傳分析,發現遺傳分化程度和采樣點海拔高度差之間存在顯著的相關性,表明海拔高度引起的遺傳隔離是產生群體間遺傳分化的主要原因。基于EST-SSR標記進行的遺傳多樣性分析,發現青藏扁蓿豆群體間遺傳距離與地理距離和海拔差異之間均存在極顯著的相關性,同時也提示由海拔差異導致的局部氣候環境的異質性可能對青藏扁蓿豆野生群體的遺傳多樣性產生影響,并使之獲得對局部極端生存環境的適應。但上述分子標記技術密度較低、多態性較低且操作復雜,具有一定的局限性。因此,為了進一步解析青藏扁蓿豆對極端環境的適應機制,評價青藏扁蓿豆種質資源,需要對青藏扁蓿豆開發更高效的分子標記類型。
單核苷酸多態性標記(single nucleotide polymorphism,SNP)作為第三代分子標記,已在群體選擇分析[8]、重要性狀基因定位[9]、人類群體擴張研究[10]以及動植物遺傳連鎖圖譜構建[11]等相關研究中廣泛應用,是研究物種遺傳變異的理想分子標記[12]。與以SSR為代表的二代分子標記相比,具有易于實現自動化分析、多態性高及密度高等優點。但SNP標記由于前期測序階段的高成本,限制了其大規模的開發利用,GBS(genotyping-by-sequencing)是基于二代測序技術分型SNP的一種簡化基因組測序技術,是一種高效、簡單、低成本的基因分型方法,對了解種質資源的遺傳背景和系統進化,研究群體遺傳多樣性及遺傳結構具有重要意義[13]。
本研究以5個群體共80份野生青藏扁蓿豆為材料,采用GBS技術測序,進而開發SNP標記,基于開發的SNP標記,初步進行遺傳多樣性及遺傳結構分析,為后續更加全面解析青藏扁蓿豆在青藏高原極端環境下的適應機制提供數據支持,同時也為青藏扁蓿豆種質資源評價奠定基礎。
1 材料與方法
1.1 材料
80份供試材料于2017年采集自青海省(表1),采樣群體海拔分布于2 462-3 311 m,每個群體采集16個個體,采集的單個個體放入信封袋用硅膠干燥保存備用。

表1 青藏扁蓿豆材料來源Table 1 Sources of M.archiducis-nicolai materials
1.2 方法
1.2.1 基因組DNA的提取及純化 取約0.05 g干葉用組織研磨儀進行研磨,利用改良的CTAB法[14]提取總DNA,提取緩沖液中加入少量的聚乙烯吡咯烷酮(PVP)粉末和β-巰基乙醇(14.4 mol/L),水合時間延長至5 h,期間每隔10 min震蕩混勻一次,使干葉細胞充分裂解,異丙醇沉淀時間延長至90 min,提高DNA產率。DNA產物經0.8%瓊脂糖凝膠電泳檢測,確認目的條帶清晰明亮且完整后,利用TIANGEN Purification Kit柱式膠回收試劑盒(增強型)進行純化,純化后的產物經過濃度及純度檢測達到測序要求后,-20℃保存備用。
1.2.2 GBS測序文庫的構建 提取的DNA樣品送往北京奧維森基因科技有限公司,參照Elshire等[15]建庫技術(取樣-酶切-接頭連接-混樣-PCR擴增-純化)建庫,首先應用限制性內切酶ApeKⅠ對基因組DNA進行酶切,加上帶有條形碼的接頭后,對每個樣品進行擴增,然后對樣品進行混合,電泳回收375-400 bp區間的DNA條帶,純化后產物用于測序,測序反應在Illumina HiSeqPE150測序平臺上進行雙末端150 bp的測序。
1.2.3 SNP開發 將測序得到的原始數據按照如下標準過濾:剔除帶接頭(adapter)的reads pair;單端read中含有N的含量超過該條read長度比例的10%時,去除此對paired reads;單端read中含有的低質量(質量值Q≤5)堿基數超過該條read長度比例的50%時,也需去除此對paired reads。最終得到Clean reads進行后續分析。因為青藏扁蓿豆尚未進行全基因組測序,所以首先選取堿基數量(176 814 300)最多的 HY_14基于 Stacks2.4[16]無參分析的流程構建擬參考基因組,參數為-m=3,-M=2,-N=2;然后通過短序列比對軟件BWA(http://bio-bwa.sourceforge.net/)將每個樣本過濾后的高質量序列與擬參考基因組比對,參數:MEM-t=4,-k=32,-M;最后根據比對結果,利用GATK3.8(https://software.broadinstitute.org/gatk/)開發 SNP位點并使用Vcftools-v0.1.13(http://vcftools.sourceforge.net/)軟件對開發的SNP位點進行以下條件的過濾:對任意一個位點,群體里至少有80%的樣本有基因型、質量值最低為20、次等位基因頻率最小為0.05并且次等位基因數量大于3,從而獲得高質量的SNP位點。使用VCFTOOLS軟件對SNP分型結果進行轉換(transition)與顛換(transversion)統計并由EXCEL繪圖。
1.2.4 遺傳數據分析 利用ADMIXTURE(http://software.genetics.ucla.edu/admixture/download.html)軟件檢測群體的遺傳結構,所得結果通過Excel進行可視化繪圖;采用GCTA(https://cnsgenomics.com/software/gcta/bin/gcta_1.93.2beta.zip)進行主成分分析(principle component analysis,PCA),并使用R 包 ggplot2[17]畫圖 ;利用 ARLEQUIN 3.5[18]軟件計算遺傳多樣性指數(Pi)、觀測和期望雜合度(HO和HE)以及遺傳分化指數(FST),進行遺傳多樣性和遺傳分化程度分析;利用GENALEX6.503[19]軟件中的Mantel test對成對的群體統計地理距離,并計算其與遺傳距離的相關性。
2 結果
2.1 測序質量
對80份青藏扁蓿豆材料的測序數據進行統計,包括5個群體的平均測序片段數,平均堿基數,群體Q30與GC含量,以及酶捕獲率。結果(表2)顯示,測序共獲得60.79 Gb數據,平均每個樣本0.76 Gb,5個群體產生的測序片段數以及堿基數存在差異,其中,最多的為湟源(HY)群體(平均測序片段數為6 638 025條,平均堿基數為0.99 Gb),最少為西寧西山(XNXS)群體(平均測序片段數為4 360 916條,平均堿基數為0.65 Gb),平均Q30≥93.07%,平均GC含量≥42.29%,數據滿足分析要求。經過低質量數據過濾后得到的平均測序片段數為3 468 857(XNXS)-5 456 407(HY),群體測序平均酶捕獲率≥98.74%,總計保留下12 796個高質量SNP位點用于后續分析。

表2 青藏扁蓿豆群體測序數據統計Table 2 Summary of sequencing data of M.archiducis-nicolai populations
2.2 SNP突變類型及數量
對SNP分型結果進行突變類型統計,結果(圖1)表明,C/T轉換類型最多(4 124個),占所有堿基突變類型的32.23%,A/G轉換類型(4 037個)占31.55%;G/C顛換類型最少(518個),占所有堿基突變類型的4.05%,A/C(1 164個)、A/T(1 777個)和G/T(1 176個)顛換類型分別占9.10%、13.89%和9.19%。轉換與顛換(Ts/Tv)之比為1.761。
2.3 遺傳結構分析
基于SNP分型數據,對來自5個群體的80份青藏扁蓿豆材料進行群體遺傳結構分析,提取K=2-5時的交叉驗證錯誤率(cross-validation error,CV error)(圖2-A),可見K從2-5交叉驗證錯誤率值逐漸增大,因此,可將K=2作為最佳K值。當K=2時,群體遺傳結構圖將青海祁連(QLCD)群體與其他群體劃分開(圖2-C)。主成分分析以得到的SNP為基礎進行分析(圖2-B),結果顯示,個體的聚類情況能夠清楚地被反映出來,親緣關系的遠近也可以由群體之間的距離反映。第一主成分(PC1)將青海祁連(QLCD)群體與其他群體區分開,這與ADMIXTURE分析的結果相一致。
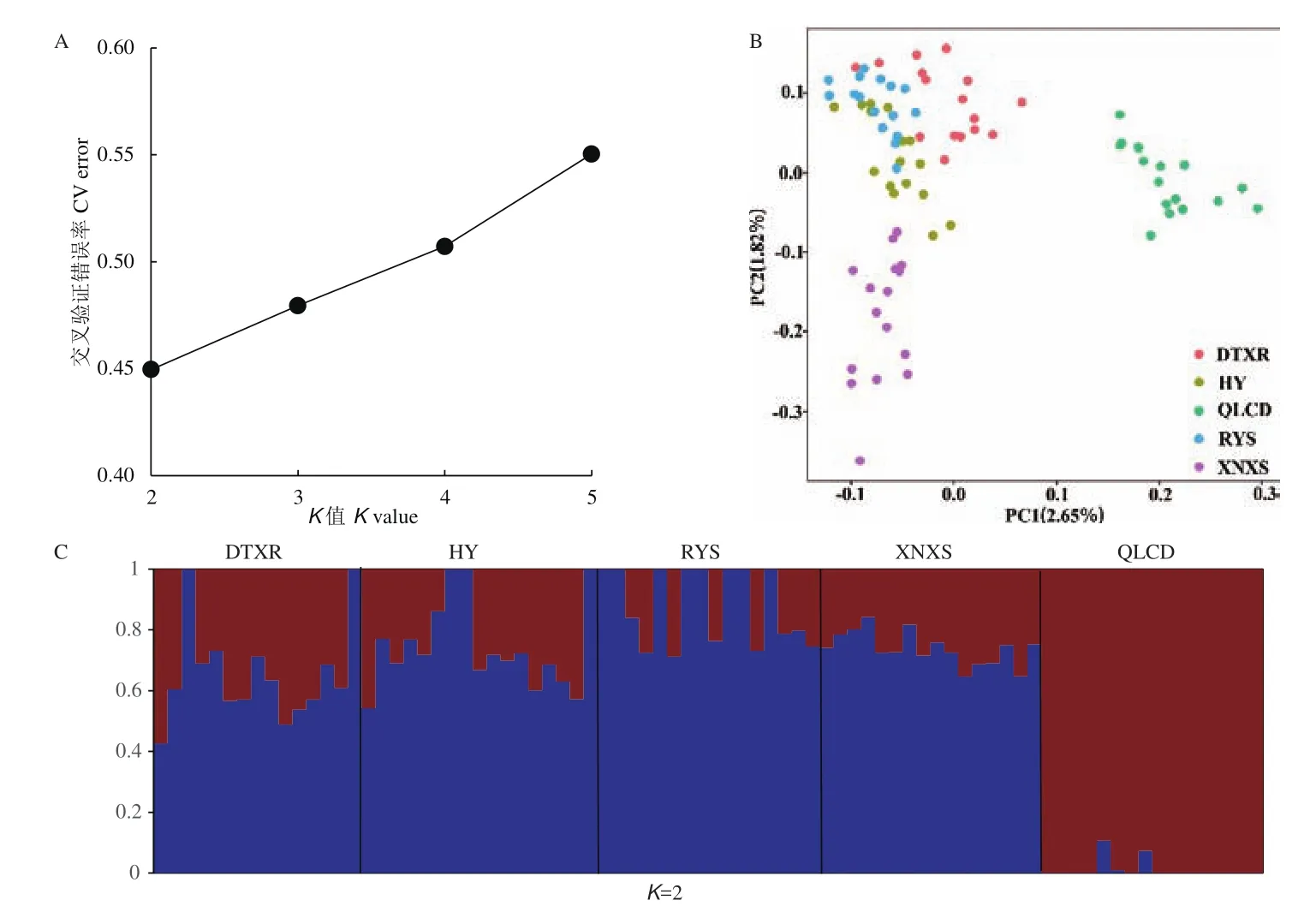
圖2 群體遺傳結構分析Fig.2 Population genetic structure of populations
利用遺傳分化指數(FST)評估青藏扁蓿豆群體間的遺傳分化程度,發現群體間的遺傳分化指數為0.009 3-0.036 9,所有群體間均存在極顯著(P<0.01)的遺傳分化(表3)。基于Mantel test分析,結果表明,青藏扁蓿豆野生群體的地理距離與遺傳距離之間存在極顯著的正相關關系(R2=0.877 6,P=0.006,圖3)。
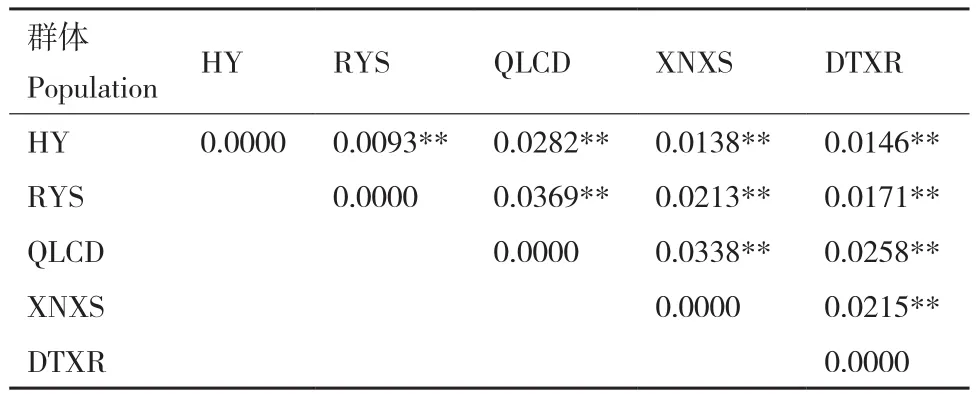
表3 群體間遺傳分化指數(FST)Table 3 Index of genetic differentiation(FST)between populations
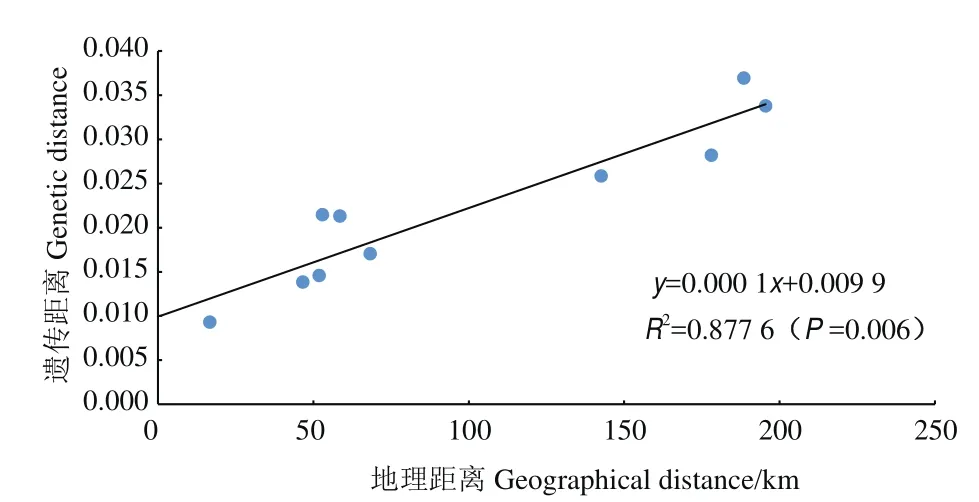
圖3 群體遺傳距離與地理距離的相關性Fig.3 Correlation between population genetic distance and geographical distance
2.4 遺傳多樣性分析
基于開發的12 796個SNP位點進行遺傳多樣性分析,發現各群體的平均等位基因數(NA)均為2;觀測雜合度(HO)在0.187 68(RYS)-0.304 36(QLCD);期望雜合度(HE)在0.201 97(RYS)-0.364 34(QLCD);遺傳多樣性指數(Pi)為0.178 32(RYS)-0.241 34(QLCD)。 表 明 日 月 山(RYS)群體具有相對最低的遺傳多樣性水平,青海祁連(QLCD)群體具有相對最高的遺傳多樣性水平(表4)。
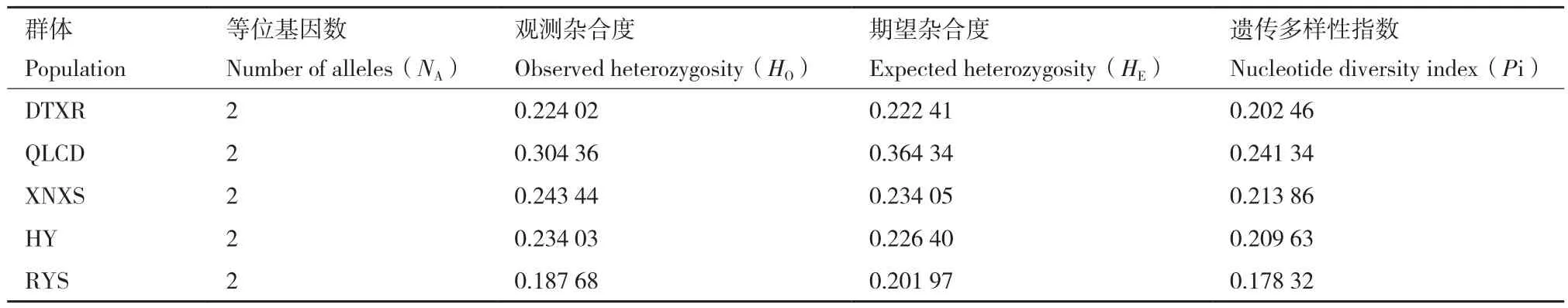
表4 青藏扁蓿豆野生群體遺傳參數統計Table 4 Statistics of genetic parameters in M.archiducis-nicolai wild populations
3 討論
簡化基因組測序技術適合于大樣本量的研究,可快速鑒定出高密度的變異,特別是SNPs變異,在物種進化、遺傳多樣性分析、全基因組關聯分析等領域中應用越來越廣泛。其中,GBS技術運用了甲基化敏感的限制性內切酶,回避了基因組主要的重復區域,使得文庫構建簡單,成本相對低廉,且有助于高通量SNPs分型技術的大規模應用。前人對煙草進行GBS測序,利用得到的SNP位點建立的聚類樹將供試材料進行了準確劃分[20]。也有研究表明,利用GBS技術對紫花苜蓿品系進行測序,并結合表型性狀進行全基因組關聯分析,可得到與黃萎病性狀顯著相關的SNP標記[21]。本研究利用GBS技術對80個青藏扁蓿豆樣本進行測序,得到了60.79 Gb的原始數據,經過挖掘并過濾最終獲得了12 796個高質量的SNP位點,用于后續遺傳多樣性和遺傳結構分析。
堿基置換突變是形成生物個體多態性和推動種群進化的根本原因之一,大部分堿基置換突變只涉及單個堿基的替換,即轉換(transition)和顛換(transversion)[22]。轉換是嘧啶互換或嘌呤互換,如C/T和A/G型SNP;顛換則是嘧啶和嘌呤相互替換,如A/C、C/G、A/T和G/T型SNP。本研究中所獲得的SNP位點堿基轉換類型占63.78%,遠高于堿基顛換(36.22%)的占比,與嚴佳文等[23]研究的火龍果堿基變異類型(轉換占63.29%,顛換占36.71%)相同。由于轉換有4種可能性,顛換有8種可能性,因此,理論上發生轉換概率與發生顛換概率的比值(Ts/Tv)應該等于0.5。但實際上,Ts/Tv值往往大于0.5,這種差異性被稱為“轉換偏差”[24-26]。本研究挖掘的SNP突變類型存在明顯的轉換型偏差現象,Ts/Tv=1.761>0.5,產生這種差異的原因可能與不同物種在進化中承受的選擇壓力有關[27]。
一個物種的進化潛力和抵御不良環境的能力既取決于種內遺傳變異的高低[28],也有賴于遺傳變異分布格局,即群體的遺傳結構[29-30]。本研究對青藏扁蓿豆野生群體的群體遺傳結構分析表明,5個青藏扁蓿豆群體劃分為2個大的類群,而之前研究并沒有將其分開[7],因此,SNP標記與EST-SSR標記相比具有更高的分辨率。群體的遺傳分化指數(FST)是衡量群體間遺傳分化程度的重要參數,可以解釋影響種群發生遺傳分化的因素[31]。本研究5個群體中,群體分化指數為0.009 3-0.036 9,且所有群體間的群體分化指數均小于0.05,根據Wright[32]對遺傳分化指數的界定,FST<0.05表明群體遺傳分化較弱,0.05<FST<0.15表明群體中等程度遺傳分化,0.15<FST<0.25表明群體較大程度遺傳分化,FST>0.25表明群體極大程度遺傳分化。因此,本研究中群體間遺傳分化較弱。此前研究顯示,來自青海卓尼、青海樂都以及內蒙古的扁蓿豆供試材料的群體分化指數為0.047 1-0.138 2,青藏扁蓿豆供試材料中的群體分化指數介于-0.055 2-0.062 4[5],表明青藏扁蓿豆群體的傳分化程度較低。本研究中的5個青藏扁蓿豆群體間的分化指數也較低,表明各群體間遺傳分化程度均較弱、親緣關系相對均較近。對青藏扁蓿豆群體遺傳距離與地理距離的相關性分析表明,地理距離與群體的遺傳分化存在極顯著(P<0.01)的相關性,與此前基于EST-SSR標記研究的結果相一致[7],推測可能與冰期后居群由避難所向研究群體擴散過程中發生的奠基者效應有關。
高原抬升,亞洲季風、亞洲內陸干旱化以及第四紀冰期等諸多因素共同作用造就了青藏高原復雜獨特的地貌環境和豐富的遺傳多樣性[33]。本研究發現,位于青藏高原北部的青海祁連(QLCD)群體具有相對較高的遺傳多樣性,而此前研究表明,來源于青藏高原東南邊緣的青藏扁蓿豆群體具有相對較高的遺傳多樣性[6-7],推測高原東南邊緣可能存在青藏扁蓿豆冰期避難所。造成這種結果的差異可能是由于采樣群體數過少,且群體分布相對集中,依據并不充分。因此,下一步可擴大采樣群體范圍來進行后續研究分析。
4 結論
運用GBS測序技術在80份野生青藏扁蓿豆材料中開發了12 796個高質量的SNPs。青海祁連(QLCD)群體的遺傳多樣性水平相對最高;5個群體被劃分為2個大的類群,此結果與主成分分析結果基本一致。另外,該青藏扁蓿豆群體間的遺傳分化程度較弱,地理距離與群體的遺傳分化程度呈現極顯著(P<0.01)的相關性。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
