MC2R基因變異致家族性糖皮質激素缺乏癥1例
2022-06-06 03:14:24袁高品張曉紅張豐豐陳婷麗
福建醫科大學學報 2022年2期
袁高品, 張曉紅, 張豐豐, 陳婷麗
家族性糖皮質激素缺乏癥(familial glucocorticoid deficiency, FGD)是一種罕見的以孤立的糖皮質激素缺乏而鹽皮質激素活性正常為特征的常染色體隱性遺傳病。該病是由于腎上腺皮質束狀帶對促腎上腺皮質激素(adrenocorticotropic hormone, ACTH)不敏感而使皮質醇水平低下,ACTH明顯增高,也稱ACTH不敏感綜合征。目前主要有3種亞型:由黑皮質素2受體(melanocortin 2 receptor, MC2R)基因致病變異引起的FGD1型(OMIM 202200);由MC2R輔助蛋白質(melanocortin 2 receptor accessory protein, MRAP)基因致病變異引起的FGD2型(OMIM 607398);由非典型的類固醇生成急性調節蛋白(steroidogenic acute regulatory protein, STAR)基因致病變異引起的FGD3型。其他報道與FGD相關的基因包括MCM4、NNT、TXNRD2、CYP11A1和SGPL1基因[1-2]。不同病因引起的FGD有不同的臨床表現,共同的臨床表現為糖皮質激素缺乏。FGD1占FGD的25%,主要表現為皮膚色素沉著、低血糖、反復感染、高膽紅素血癥和高身材[2-3],也有一些面容異常[3-5],如前額突出、眼間距過寬、內眥贅皮、寬鼻梁和其他部位畸形的報道,如小而尖的手指、通貫掌。本研究報告1例FGD1型病例,具體如下。
1 病例介紹
1.1 臨床資料 患兒,女,出生后1 d出現發紺、氣促,心率及血氧下降,予胸外心臟按壓、氣囊加壓給氧治療后心率恢復,血氧欠佳,氣管插管下轉診筆者醫院。患兒系G1P1孕41周行剖宮產娩出,出生體質量2 920 g,身長50 cm,出生后1、5和10 min Apgar 評分分別為9、10和10分。父母體健,非近親婚配,家族中無類似癥狀及遺傳性疾病病史。體格檢查:血壓48/31 mmHg(1 mmHg=133.3 Pa),神志清,全身皮膚黏膜及外陰色素沉著,前額突出,眼距寬,甲狀腺無腫大,心、肺、腹未見明顯異常,雙手通貫掌,陰蒂無肥大。實驗室檢查腎功能、心肌酶、血脂均正常,總膽紅素152.8 μmol/L;血氣分析:pH值為7.201、BE為-10.4 mmol/L、碳酸氫根17.5 mmol/L、鉀3.84 mmol/L、鈉130.4 mmol/L、葡萄糖2.01 mmol/L;血串聯質譜及尿有機酸分析未見明顯異常;胸部正位片:呼吸窘迫綜合征;心臟彩超及顱腦MRI顯示未見明顯異常;染色體46,XX。入院后,患兒反復出現頑固性低血壓(35/20 mmHg),予補液,補充白蛋白,多巴胺、多巴酚丁胺聯合去甲腎上腺素升血壓處理,血壓仍低,加用氫化可的松維持血壓,血壓穩定后停用,3 d后出現低鈉低氯血癥(鈉133.3 mmol/L,氯97.4 mmol/L)伴高鉀血癥(鉀6.0 mmol/L)。甲狀腺功能檢查顯示,促甲狀腺激素(thyroid stimulating hormone, TSH)為25.54 μIU/mL,FT3和FT4正常;ACTH為1 250 pg/mL;皮質醇<0.8 μg/dL,睪酮<7 ng/dL;孕酮<0.21 ng/mL;17α羥孕酮為0.35 nmol/L,雄烯二酮<0.3 ng/mL。甲狀腺及腎上腺彩超正常,提示原發性腎上腺功能不全(primary adrenocortical insufficiency, PAI)。加用氫化可的松[27 mg/(m2·d)]及左甲狀腺素治療。患兒PAI診斷成立,建議完善基因檢測,明確病因。
1.2 醫學外顯子組測序結果及分析 在簽署知情同意書后,采集患兒及其父母的外周血樣2 mL(EDTA抗凝),送廈門基源醫學檢驗實驗室進行全外顯子組基因檢測。從先證者外周血白細胞中提取全基因組DNA,進行DNA建庫和捕獲,并進行150 bp 雙末端測序。測序原始數據經生物信息分析處理后,采用Burrows-Wheeler Alignment tool (BWA)軟件進行序列比對,并用GATK軟件(https://software.broadinstitute.org/gatk/)分析變異。采用Annovar軟件進行變異注釋(注釋信息包括染色體起始和終止位置,參考等位基因,替代等位基因,基因功能,千人基因組和ExAC等數據庫人群頻率SIFT/Provean/ Mutation taster等蛋白功能預測軟件結果)。結合先證者的臨床表型,參考美國醫學遺傳學和基因組學學會(American College of Medical Genetics and Genomics, ACMG)遺傳變異分類標準與指南[6-7]對檢出變異進行解讀,結果顯示患兒MC2R基因2號外顯子存在2個復合雜合變異:NM_001291911(MC2R):c.145G>A(p.Val49Met),NM_001291911(MC2R):c.536C>A(p.Thr179Lys)(圖1),分別遺傳自父母。其中c.145G>A(p.Val49Met)變異導致第49位氨基酸由纈氨酸變為甲硫氨酸。該變異在千人基因組數據庫中未收錄,在ExAC等數據庫中等位基因頻率為0.000 1,在In-House數據庫MAF頻率為0.000 014 6,生物信息學軟件預測為有害(SIFT: Damaging, Polyphen2_HDIV:possibly damaging, Polyphen2_HVAR: Possibly damaging, Mutation taster: disease_causing),蛋白結構預測可能有害(Polyphen2, SIFT, Mutation taster),軟件預測該核苷酸具有高度保守性(GERP, phyloP, phastCons)。根據ACMG指南,判定該變異為可能致病性變異(致病性證據等級為PM1-PM3-PM2_supporting-PP3)。c.536C>A(p.Thr179Lys)變異導致第179位氨基酸由蘇氨酸變為賴氨酸。該變異在千人基因組、ExAC、In-House等數據庫中未收錄,生物信息學軟件預測為有害(SIFT: damaging; Polyphen2_HDIV: probably damaging; Polyphen2_HVAR: probably damaging; Mutation taster: disease_causing; Provean: deleterious),蛋白結構預測可能有害(Provean, SIFT, Mutation taster),軟件預測該核苷酸具有高度保守性(GERP, phyloP, phastCons)。根據ACMG指南判定該變異為可能致病性變異(致病性證據等級為PM1-PM2_supporting-PM3-PP3)。
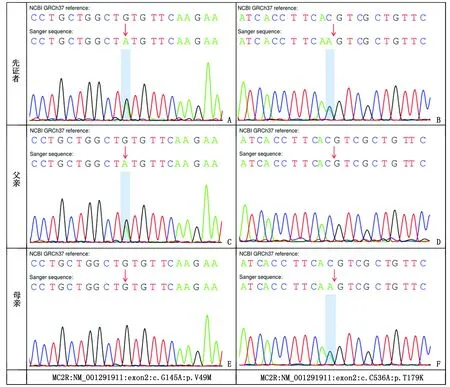
A、B:患兒分別攜帶c.145G>A(p.Val49Met)和c.536C>A(p.Thr179Lys)雜合變異;C、D:患兒父親攜帶c.145G>A(p.Val49Met)雜合變異,未攜帶c.536C>A(p.Thr179Lys)雜合變異;E、F:患兒母親未攜帶c.145G>A(p.Val49Met)雜合變異,攜帶c.536C>A(p.Thr179Lys)雜合變異。圖1 先證者及其父母MC2R基因測序結果Fig.1 MC2R gene sequencing results of the proband and her parents
1.3 隨訪情況 出院后門診隨診,監測電解質正常,ACTH 1 250 pg/mL,皮質醇<0.8 μg/dL,予氫化可的松逐漸減至生理需要量。甲狀腺功能及神經系統發育正常。嘗試停用左甲狀腺素,停藥2周后,TSH升至6.85 μIU/mL,1個月時升至9.344 μIU/mL,考慮TSH進行性升高,再次予左甲狀腺素治療。現1歲5個月,身長85 cm,體質量10.4 kg,頭圍45.5 cm,胰島素樣生長因子1(insulin-like growth factor 1, IGF-1)、胰島素樣生長因子結合蛋白(insulin-like growth factor binding protein 3,IGFBP-3)正常,立位醛固酮為275.17 pg/mL,腎素活性為22.15 ng/(mL·h)。
2 討 論
本研究報道了1例MC2R基因致病變異所致的FGD1新生兒,有助于提高臨床醫師對FGD1的認識。該患兒出生后即出現皮膚黏膜色素沉著,頑固性低血壓,伴有低鈉、高鉀血癥,皮質醇降低,ACTH顯著升高,符合PAI的診斷。體見輕度的面部異常,表現為前額突出、眼距寬,伴有雙手通貫掌,實驗室檢查存在甲狀腺功能異常,予小劑量左甲狀腺素治療,甲狀腺功能維持正常,曾嘗試停左甲狀腺素,停藥后TSH進行性升高,再次予左甲狀腺素治療。門診隨診監測身長,存在生長加速現象。
FGD1是PAI的一種特殊類型,較罕見,發病率不明確。我國于2019年報道首例FGD1,至今共報道3例[4,8-9]。典型的臨床表現為色素沉著、身材高大。隨著報道病例的增加,FGD1的臨床表型也不斷增多。FGD1可能伴有其他的內分泌異常[4-5,10-15],最常見的是甲狀腺功能異常[2,4,10,16],多表現為暫時性單純TSH升高,予左甲狀腺素短期治療或未經治療后恢復正常。部分FGD1患者存在部分或完全性激素缺乏[4,11,13],如17α羥孕酮、雄烯二酮、脫氫表雄酮、睪酮、孕酮和硫酸脫氫表雄酮。FGD1患者可能出現陰毛發育延遲,但其他性特征不受影響。FGD1臨床表現為單純糖皮質激素缺乏而鹽皮質激素活性正常,可見短暫的鹽丟失或低鈉血癥,但真正的鹽皮質激素缺乏非常少見[12,14-15]。CHAN等[14]報道6例FGD1出現鹽皮質激素不足,均為純合無義或移碼變異,提示MC2R基因嚴重或純合變異可干擾腎素-血管緊張素-醛固酮系統,可能需要短暫的鹽皮質激素治療,但不會出現長期的鹽皮質激素缺乏。另外,典型的異常面容有助于FGD1的診斷,患者可有面容異常或其他部位畸形。SLAVOTINEK等[5]首次報道1例具有R146H純合變異的FGD1患兒具有寬鼻梁、內眥贅皮和小而尖的手指。后續研究也報道患兒具有前額突出、眼間距過寬、內眥贅皮或通貫掌等異常面容[3-4];部分合并肌無力[10]、運動發育遲緩[17]等。
本研究患兒查體有前額突出、眼距寬、雙手通貫掌,與文獻[3-4]報道的病例面容相似。實驗室檢查17α羥孕酮、雄烯二酮降低,結合患兒月齡處于小青春期,睪酮、孕酮相對偏低,提示存在性激素缺乏。病程中有低鈉血癥,予糖皮質激素替代,監測電解質正常,查醛固酮水平正常,未提示鹽皮質激素缺乏,與上述文獻報道一致。本研究患兒已1歲5個月,仍需左甲狀腺素治療,尚需進一步隨診以明確甲狀腺功能異常是否也為暫時性。
FGD1出生時身長正常,診斷時身材高大[11-12,18],提示生長加速發生在出生后。本研究中患兒出生時身長正常,新生兒期即予生理替代量的氫化可的松治療,隨訪至1歲5個月,身長位于同年齡同性別1.5個標準差,提示在生理替代量氧化可的松治療的情況下,患兒仍存在生長加速現象。IGF-1和IGFBP-3 水平正常,提示生長加速與生長激素/IGF-1軸無關,與文獻報道一致[3,5,13]。有學者認為,FGD1患兒身材高大與長時間暴露于ACTH和慢性糖皮質激素缺乏有關[18],認為FGD1患兒經糖皮質激素替代治療,ACTH顯著下降,晚期生長速率正常化[3,19]。文獻中使用的糖皮激素劑量較大,身高正常化可能與大量的糖皮質激素抑制生長相關。也有學者認為,高身材可能是ACTH激活骨和軟骨生長板中表達的其他黑皮質素受體并刺激生長,或者刺激雌激素的合成促進生長[20]。
FGD1呈常染色體隱性遺傳,本例患兒父母表型正常,Sanger測序驗證患兒檢出的2個MC2R基因致病變異分別遺傳自父母,呈反式排列,與FGD1的遺傳模式相符合。
MC2R是7個跨膜的G蛋白偶聯受體,在腎上腺皮質高表達,與ACTH結合導致細胞內環磷酸腺苷增加和蛋白激酶A激活,控制著糖皮質激素的分泌[7]。MC2R基因突變導致FGD1的潛在機制可能是由于受體從內質網到細胞表面轉運障礙,細胞表面受體表達減少,少數轉運至細胞表面的受體位于MC2R的胞外結構域,導致配體與受體結合失敗,引起糖皮質激素分泌缺陷[21-22]。本研究中,c.145G>A(p.Val49Met)變異在千人基因組數據庫中未檢出,在ExAC數據庫中等位基因頻率極低,采用SIFT、Polyphen2_HDIV、Polyphen2_HVAR、Mutation taster等生物信息學軟件預測為對蛋白功能有害的變異,該變異曾在1個FGD1男孩中檢出。p.Val49Met變異位于第1個跨膜結構域和第1個胞內環的交界處。根據分子模型,Val49Met變異導致第1個跨膜結構域的結構改變,以及第一跨膜結構域和第7個跨膜結構域潛在新的相互作用,可能導致信號轉導丟失,進而導致糖皮質激素分泌缺陷,且纈氨酸在不同物種之間高度保守。在60個不相關的健康對照者中未發現該變異,也提示該變異可能在MC2R功能中發揮重要作用[10]。c.536C>A(p.Thr179Lys)變異在千人基因組、ExAC等數據庫中均未檢出,采用SIFT、Polyphen2_HDIV、Polyphen2_HVAR、Mutation taster等生物信息學軟件預測為對蛋白功能有害的變異,蛋白結構預測可能有害(Polyphen2, SIFT, Mutation taster)。患兒臨床表型及遺傳方式均與FGD1相符,根據ACMG指南,c.145G>A(p.Val49Met)和c.536C>A(p.Thr179Lys)均為可能致病性變異。因此,認為本例攜帶的MC2R基因c.145G>A(p.Val49Met)和c.536C>A(p.Thr179Lys)變異是致病變異,尚需進一步的功能研究來驗證此變異對MC2R蛋白功能的影響。
綜上所述,本研究報道1例新生兒發病的FGD1病例,具有特殊的面部特征,伴甲狀腺功能異常;新發現一個MC2R基因致病變異——c.536C>A(p.Thr179Lys),豐富了MC2R基因變異譜。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
中國科技論壇(2017年7期)2017-07-25 08:49:53
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
