MoS2/Ru異質結構的制備及其電催化析氫反應性能
2022-05-23 04:53:38馬海霞王太和趙玉潔李嘉辰
材料工程 2022年4期
馬海霞,王太和,趙玉潔,王 旭,李嘉辰,2
(1 西北大學 化工學院,西安 710069;2 西北大學 化學與材料科學學院,西安 710127)
氫能由于其具有高能量密度和清潔無污染等特性并且可通過可再生能源而獲取被認為是可以替代化石能源的最佳替代品,因此能夠有效緩解全球氣候變暖以及環境污染等問題[1-2]。電催化水分解制氫是最清潔的工業制氫技術,由于采用高昂的鉑(Pt)催化劑以達到降低反應能耗的目的,不可避免地提高了電水解制氫成本,在一定程度上限制了這種制氫技術的發展。鑒于此,許多研究者致力于設計低成本、高活性與穩定性的非貴金屬催化劑替代貴金屬Pt催化劑從而降低反應能耗的同時降低制氫成本[3-4]。
近些年來,二維層狀二硫化鉬(2D MoS2)由于其結構類似于自然界中的光催化氫化酶和在酸性或堿性條件下的理論氫吸附能更接近于熱中性(ΔG=0)而作為氫析出反應(HER)催化劑被廣泛研究[5-6]。通過理論計算得知,電子密度更趨近于MoS2中的Mo邊位點,從而質子(H+)更容易在Mo活性位點上形成Mo—H鍵[7]。對于堿性HER,其反應機理存在兩種不同的路徑:Volmer-Tafel或者Volmer-Heyrovsky路徑,包括水分子中HO—H鍵的斷裂(Volmer步驟)而后進行電化學脫附H2(Heyrovsky步驟)或化學脫附H2(Tafel步驟)。在此過程中,Volmer步驟在堿性HER過程中是速率決定步驟[8]。MoS2在堿性環境中被證明是能夠促進Heyrovsky步驟,但是MoS2表面對OH-吸附能力較強,這導致了Volmer步驟動力學反應速率較低,致使在堿性環境下的反應速率相比于酸性環境下降了2~3個數量級[9-11]。
近年來,許多研究者致力于如何提高MoS2在堿性條件下的HER活性而做了大量研究,包括陰陽離子摻雜[12]、相轉變(2H相轉變為1T相)[13]、構建表面缺陷[14]、構建異質結構[3,15]等。其中,將MoS2與能夠促進堿性Volmer步驟的物質構建異質結構能夠有效提高HER活性。Subbaraman等[16]最早證明了Ni(OH)2的邊位點在Pt表面能夠促進堿性HER中Volmer水分子的吸附/裂解,形成的氫中間體(Hads)轉移到臨近Pt位點析出氫氣。根據這種策略,Hu等[17]將NiCo-LDH與MoS2進行復合構建MoS2/NiCo-LDH異質結構,在1.0 mol·L-1KOH條件下電流密度-10 mA·cm-2的過電位為78 mV。隨后,許多研究者將金屬氫氧化物如Ni(OH)2[10],Co(OH)2[18],CoO[19]等金屬氧化物/氫氧化物作為Volmer步驟的促進體與MoS2結合,表現出了優異的HER活性。然而,這些非貴金屬催化劑仍然不能達到Pt的活性。因此,將貴金屬與MoS2結合并且調節ΔGH能夠有效提高HER活性,同時將貴金屬納米化甚至單原子化以最大程度上提高貴金屬質量活性和降低成本。最近,有研究者將Pt、鈀(Pd)、銠(Rh)等貴金屬與MoS2進行結合作為堿性HER催化劑,表現出類Pt的性質[20-22]。而在Pt族金屬中,釕(Ru)是成本最低的金屬,并且Ru—H與Pt—H的鍵能相近(≈65 kcal/mol)[23]。此外,Ru被證明能夠誘導局域結構極化,有效降低Volmer活化能壘,并且能夠優化ΔGH[24]。因此,利用Ru代替金屬氧化物或其他貴金屬與MoS2復合可望在極大程度上提高其堿性HER活性。
本工作通過兩步水熱法合成具有高分散性超小Ru納米顆粒負載在MoS2納米片并原位生長在三維導電碳布上,形成CC@MoS2/RuNPs異質結構電催化劑。對其進行結構和形貌的表征以及堿性HER電化學活性測試,考察了不同RuNPs負載量對HER活性的影響,優化出最佳MoS2/Ru配比。并且測試了該催化劑在長時間循環后的形貌和結構穩定性。最終討論了MoS2與Ru在堿性HER過程中的協同催化作用機制。
1 實驗材料與方法
1.1 CC@MoS2納米片(NSs)的制備
在進行水熱反應之前,將裁剪好的CC(1 cm×3 cm)用3.0 mol·L-1HCl、丙酮和無水乙醇分別清洗數次以去除CC表面的油漬和雜質,清洗完后將其浸泡在無水乙醇中備用。稱取1.0 mmol Na2MoO4·2H2O和4.0 mmol硫脲溶解在15 mL去離子水中,攪拌10 min后倒入容量為25 mL聚四氟乙烯內膽的不銹鋼水熱釜中。將溫度調至220 ℃并保溫24 h,隨后將CC@MoS2NSs用去離子水和乙醇清洗并在60 ℃下干燥即可得到CC@MoS2納米片(NSs)。
1.2 CC@MoS2/RuNPs的制備
將上述制得的CC@MoS2NSs(1 cm×1 cm)浸入Ru3+的乙醇溶液中,該溶液由2.5 mL RuCl3·xH2O水溶液(10 mg/mL)倒入15 mL乙醇并攪拌10 min制得。將乙醇溶液倒入容量為25 mL聚四氟乙烯內膽的不銹鋼水熱釜中,溫度調至180 ℃并保溫6 h。隨后取出CC@MoS2/RuNPs并用去離子水和乙醇清洗后在60 ℃下干燥。為了研究不同RuNPs負載量對性能的影響,可通過控制Ru3+溶液的量調節MoS2與RuNPs的比例。本工作將RuCl3·xH2O水溶液的添加量為0.2,0.4,0.8,1.0,1.5,2.0,2.5,3.0 mL和3.5 mL分別記作CC@MoS2/Ru-0.2,CC@MoS2/Ru-0.4,CC@MoS2/Ru-0.8,CC@MoS2/Ru-1.0,CC@MoS2/Ru-1.5,CC@MoS2/Ru-2.0,CC@MoS2/Ru-2.5,CC@MoS2/Ru-3.0和CC@MoS2/Ru-3.5。CC@MoS2/RuNPs的制備流程圖如圖1所示。
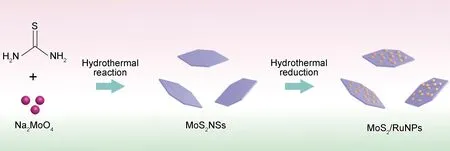
圖1 MoS2/RuNPs的制備流程圖Fig.1 Schematic illustration of preparation procedures for MoS2/RuNPs
1.3 材料表征
樣品的結構和形貌表征分別采用X射線粉末衍射儀(XRD,Bruker D8 Advance)、掃描電子顯微鏡(SEM,Hitachi, SU8010)、透射電子顯微鏡(TEM,FEI Talos F200X)、X射線光電子能譜(XPS,Thermo Scientific Nexsa),粒徑分布圖是由Nano Measurer 1.2軟件進行分析繪制。
1.4 電化學測試
本工作的電化學測試在CHI760E電化學工作站進行。采用三電極體系進行堿性HER測試,其中所制催化劑為集成工作電極,無需任何黏結劑和導電劑。石磨棒和Hg/HgO(1.0 mol·L-1KOH)分別為對電極和參比電極;電解液體系為1.0 mol·L-1KOH。本工作中所有電位通過公式ERHE=EHg/HgO+0.059×pH+0.098換算為可逆氫電極(RHE)。線性掃描伏安法(LSV)測試的掃描速度為2.0 mV/s。循環伏安法(CV)測試在非法拉第電位區間,通過不同掃描速度v(5,10,25,50,100 mV/s)和雙電層電流(ic)的線性關系(見式(1))[25]計算出雙電層電容(Cdl),進而判斷不同材料的電化學表面積(ECSA)的大小。穩定性測試可通過考察2000周次CV前后的LSV重合度,或通過在恒定電流密度(-10 mA·cm-2)下電位隨時間變化趨勢(計時電位滴定法,CP)進行判斷。
ic=vCdl
(1)
2 結果與分析
2.1 結構與形貌表征
圖2為CC@MoS2和CC@MoS2/RuNPs的XRD譜圖,可以看出CC@MoS2的衍射峰和MoS2標準卡片(JCPDS No.37-1492)完全吻合,說明所制備的樣品不存在任何雜相[26-27]。在24°左右出現一個寬峰,對應三維基底CC特征峰。在14.4°位置說明MoS2(002)晶面為MoS2層間距。另外,在29°,33.5°,39.5°,44.2°,49.8°和58.3°位置出現明顯衍射峰,并與(004),(101),(103),(006),(105)和(110)晶面相對應。與CC@MoS2相比,CC@MoS2/Ru基本保留了MoS2的特征峰,但是沒有出現任何Ru金屬的特征峰,說明在MoS2納米片表面沉積的Ru的含量較少并且顆粒粒徑較小,同時未檢測出其他Ru氧化物的信號,說明所制備的CC@MoS2/Ru純度較高。

圖2 CC@MoS2和CC@MoS2/RuNPs的XRD譜圖Fig.2 XRD patterns of CC@MoS2 and CC@MoS2/RuNPs
圖3為CC@MoS2NSs和CC@MoS2/RuNPs的SEM圖。從圖3(a-1),(a-2)中可以看出,MoS2納米片均勻生長在三維導電碳布上。在CC@MoS2NSs表面沉積RuNPs后,基本保持MoS2納米片狀形貌,表明CC@MoS2/RuNPs具備良好的結構穩定性(圖3(b-1),(b-2))。這種2D層狀MoS2結構能夠暴露更多的表面活性位點,可以充分分散RuNPs,防止團聚。而3D導電碳布基底具有低成本和高比表面積等特性,不僅可作為理想的催化劑負載基體,也可有效提高Ru利用率[28]。

圖3 樣品的SEM圖(a)CC@MoS2NSs;(b)CC@MoS2/RuNPs;(1)低倍;(2)高倍Fig.3 SEM images of samples(a)CC@MoS2NSs;(b)CC@MoS2/RuNPs;(1)low magnification;(2)high magnification
為了進一步分析CC@MoS2/RuNPs的微觀結構,本工作中對MoS2/RuNPs進行了TEM以及高分辨HRTEM的表征。圖4(a)為MoS2/RuNPs的TEM圖片,納米片表面出現多層MoS2的褶皺,但是沒有發現明顯的顆粒狀Ru,說明其粒徑較小。進一步提高TEM分辨率能夠看到MoS2NSs表面均勻分散了大量Ru顆粒(圖4(b)),沒有出現明顯的Ru顆粒團聚,證實了上述XRD的分析。另外,通過粒徑分布圖分析得知RuNPs的平均粒徑為2.5 nm(圖4(b)插圖)。圖4(c)為MoS2/RuNPs的HRTEM圖片,通過分析MoS2的晶格間距為0.62 nm,對應MoS2(002)層間距[29]。而Ru的分散區域未出現MoS2(002)晶格,說明RuNPs分散在單層MoS2表面。這種單層結構能夠暴露更多活性位點,MoS2/Ru界面具有高電活性,促進MoS2與Ru之間的電荷重新分布,表現出更加優異的催化活性[8,30]。

圖4 CC@MoS2/RuNPs的TEM圖(a)低分辨率;(b)高分辨率和粒徑分布圖;(c)HRTEM圖Fig.4 TEM images of CC@MoS2/RuNPs (a)low resolution;(b)high resolution and particle size distribution map;(c)HRTEM image
采用XPS分析CC@MoS2/Ru中元素組成以及各元素價態。圖5(a)為CC@MoS2/Ru全譜圖,其中含有Mo,S,Ru,C以及O元素,C和O元素來源于空氣污染的C源和三維CC基底,而O元素來源于材料表面不可避免的部分氧化。因此,CC@MoS2/Ru中不含其他雜元素。圖5(b)是Mo3d高分辨XPS譜圖,在226.7 eV為MoS2中S2s信號,229.5 eV和232.5 eV處的特征峰分別為MoS2中Mo4+的3d5/2和3d3/2自旋軌道。在235.3 eV處為CC@MoS2/Ru表面部分氧化形成Mo6+[31]。在圖5(c)中S2p圖譜中,其中在162.3 eV和163.4 eV分別表示MoS2中Mo—S鍵的2p3/2和2p1/2自旋軌道。在168.7 eV處的特征峰為MoS2表面氧化形成的S—O化合物[32]。圖5(d)為CC@MoS2/Ru中Ru3d高分辨XPS圖譜,可以看到結合能為280.3 eV處為Ru3d5/2自旋軌道,與Ru0相對應,表明CC@MoS2/Ru中Ru的存在形式為金屬態而非氧化態。另外,284.6 eV為C1s特征峰,主要來源于導電碳基底[33]。

圖5 CC@MoS2/Ru的XPS譜圖 (a)全譜圖;(b)Mo3d;(c)S2p;(d)Ru3dFig.5 XPS spectra of CC@MoS2/Ru (a)survey spectrum;(b)Mo3d;(c)S2p;(d)Ru3d
2.2 堿性HER電化學測試
通過對CC@MoS2/Ru進行LSV極化曲線測試可以評價材料的HER催化活性。如圖6(a)所示,不同Ru的負載量對HER活性有顯著影響。在電流密度為-10 mA·cm-2下CC@MoS2的過電位為150.8 mV,通過負載不同RuNPs的量可調控CC@MoS2/Ru的HER活性,當RuCl3·xH2O溶液的添加量為2.5 mL時,催化活性達到最優。過電位在71.3 mV和188.6 mV時就可分別驅動-10 mA·cm-2和-100 mA·cm-2的電流密度。這種優異的活性明顯高于最近發表的Ru或Mo基電催化劑(圖6(b))。但是在這里存在一個活性峰值,在RuCl3·xH2O溶液添加量超過2.5 mL,如達到3.0 mL和3.5 mL時,CC@MoS2/Ru的活性顯著降低,其原因有可能是MoS2表面負載過量RuNPs導致團聚現象,顆粒團聚會導致表面活性位點數減少,很大程度上影響催化活性。
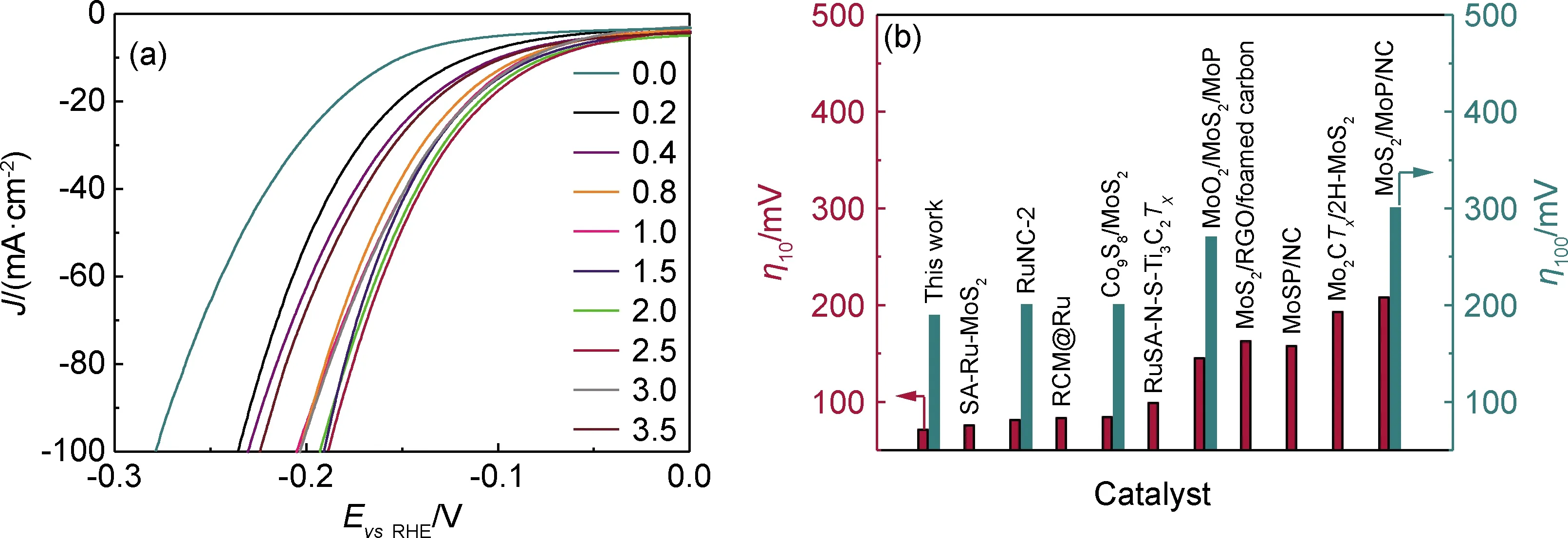
圖6 不同RuNPs負載量的CC@MoS2/Ru的LSV極化曲線(a)和在-10 mA·cm-2和-100 mA·cm-2下與最近報道的Mo或Ru基電催化劑的堿性HER性能對比圖(b)Fig.6 LSV curves of CC@MoS2/Ru with different Ru mass loading(a) and comparison of the alkaline HER performance of CC@MoS2/Ru-2.5 with those of recently reported Ru- or Mo-based electrocatalysts at -10 mA·cm-2 and -100 mA·cm-2(b)
為了研究CC@MoS2/Ru與CC@MoS2的HER動力學速率變化,通過LSV曲線和式(2)[34]計算出CC@MoS2/Ru-2.5和CC@MoS2的Tafel斜率。
η=A×lg(j/j0)
(2)
式中:η為過電位;j為電流密度;j0代表交換電流密度;A代表Tafel斜率。如圖7所示,CC@MoS2/Ru-2.5的Tafel斜率為104.8 mV·dec-1,低于CC@MoS2(114.6 mV·dec-1),說明MoS2表面負載RuNPs后會提高表/界面反應動力學速率,提高HER反應速率。因此,CC@MoS2/Ru的Tafel斜率降低表明在此異質結構中,由于存在豐富的MoS2/Ru界面,提高了堿性HER過程中Volmer-Heyrovsky反應速率,從根本上提高了堿性HER的本征活性。

圖7 CC@MoS2/Ru-2.5和CC@MoS2的Tafel曲線Fig.7 Tafel plots of CC@MoS2/Ru-2.5 and CC@MoS2
除了本征活性的影響,ECSA是另一個提高HER活性的重要因素,本工作通過測試Cdl電容值判斷CC@MoS2/Ru-2.5和CC@MoS2的ECSA差異。如圖8(a),(b)所示,通過測試非法拉第電位區間在不同掃描速度下的CV曲線,即可通過式(1)擬合出CC@MoS2/Ru-2.5和CC@MoS2的Cdl電容值。如圖8(c)所示,CC@MoS2/Ru-2.5的Cdl為102.2 mF·cm-2,遠高于CC@MoS2(74.4 mF·cm-2)。結果表明,RuNPs的負載可提高CC@MoS2的ECSA,能夠暴露更多的活性位點作為催化位點,提高CC@MoS2/Ru-2.5的HER活性。
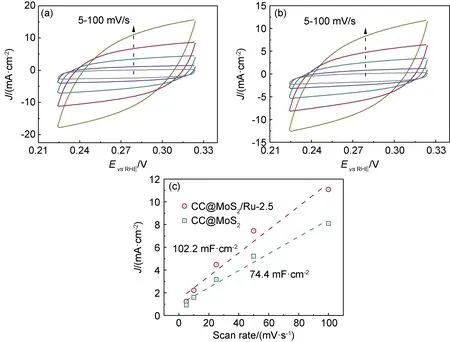
圖8 CC@MoS2/Ru-2.5(a),CC@MoS2(b)在不同掃描速度下的CV曲線以及CC@MoS2/Ru-2.5,CC@MoS2的電流密度-掃描速度關系圖(c)Fig.8 CV curves of CC@MoS2/Ru-2.5(a),CC@MoS2(b) over different scan rates and current density as a function of scan rate for CC@MoS2/Ru-2.5 and CC@MoS2 (c)
催化劑的穩定性是評判催化劑好壞的重要指標。在本工作中,采用多次CV循環前后LSV重合程度來判斷CC@MoS2/Ru-2.5的穩定性好壞。如圖9(a)所示,在2000 CV后CC@MoS2/Ru-2.5的LSV曲線與初始曲線重合度較好,說明沒有明顯的性能衰減。為了進一步證明CC@MoS2/Ru-2.5在長時間催化反應后的穩定性,本工作通過在恒定電流密度下測定過電位隨時間的變化趨勢。如圖9(b)所示,在-10 mA·cm-2電流密度下,經過至少35 h的持續催化反應后過電位變化仍不明顯,說明CC@MoS2/Ru-2.5具有優異的催化穩定性。其原因主要是MoS2/Ru原位生長在CC三維基底上,具有較強機械穩定性。另外,MoS2/Ru的三維納米片交錯排列的結構能緩解“氣體屏蔽效應”,促進H2的溢出而不會堵塞活性位點,影響后續的HER催化反應[11,23]。

圖9 CC@MoS2/Ru-2.5的穩定性測試(a)初始和2000 CV循環后的LSV曲線;(b)在-10 mA·cm-2電流密度下的CP曲線Fig.9 Stability test of CC@MoS2/Ru-2.5(a)LSV curves before and after 2000 CV test;(b)CP curve at a current density of -10 mA·cm-2
2.3 協同催化機理分析
結合實驗結果能夠看出CC@MoS2/Ru的堿性HER相比于CC@MoS2有明顯提高,說明RuNPs的復合能夠提高MoS2的本征活性。其原因主要是Ru的加入誘導Ru/MoS2的界面進行電荷重新分布,界面附近的Ru位點由于局部結構極化而積累更多的電子,從而降低Volmer水裂解的能量勢壘,解決了MoS2在堿性HER過程中的第一步水裂解動力學緩慢的問題[24]。如圖10所示,在MoS2/Ru異質結構中的堿性HER反應機理為:水分子優先在Ru位點上進行Volmer水吸附并且裂解為OH-和Hads,隨后產生的Hads進行Heyrovsky電化學脫附形成氫氣。因此,Ru與MoS2兩者的復合充分體現了協同催化作用機制。

圖10 MoS2/Ru界面的堿性HER機理示意圖Fig.10 Schematic illustration of alkaline HER mechanism on the interfaces of MoS2/Ru
3 結論
(1)通過簡單溫和的兩步水熱法制備出了新穎的CC@MoS2/Ru三維集成催化劑,表現出優異的堿性HER催化活性和穩定性。在電流密度為-10 mA·cm-2下的過電位僅為71.3 mV,相比于CC@MoS2(150.8 mV)過電位有明顯降低。在恒定電流密度下能保持至少35 h沒有明顯衰減,主要原因為CC@MoS2/Ru牢固的三維集成電極和穩定的形貌及結構,消除氣體屏蔽效應,促進H2的排出。
(2)對比不同RuNPs負載量的CC@MoS2/Ru催化活性,CC@MoS2/Ru-2.5的活性最佳,而過多負載RuNPs會降低CC@MoS2/Ru的催化活性,其主要原因是高負載量會導致RuNPs的團聚,減少活性位點的暴露。CC@MoS2/Ru優異的堿性HER活性主要來源于RuNPs的復合誘導Ru/MoS2的界面進行電荷重新分布,提高MoS2/Ru的Volmer反應速率,從根本上解決了MoS2在堿性HER活性較差的問題,有望成為替代商業Pt/C的高效穩定氫析出催化劑。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
