一測多評法測定當歸丸(濃縮丸)中3種成分的含量
2022-04-13 02:00:04樊磊磊徐金玲婁玉霞閆洪建
食品與藥品 2022年2期
樊磊磊,徐金玲,婁玉霞,閆洪建
(1. 河南省食品藥品檢驗所 國家藥品監督管理局中藥材及飲片質量控制重點實驗室,河南 鄭州 450018;2.河南中醫藥大學 藥學院,河南 鄭州 450018;3. 仲景宛西制藥股份有限公司,河南 西峽 474550)
中成藥當歸丸(濃縮丸)現收載于《衛生部中藥成方制劑第十七冊》,其功能與主治為補血活血,調經止痛,用于血虛所致的月經不調,行經腹痛[1-2]。當歸的“補血”作用主要作用于造血系統,通過刺激與造血相關的細胞、分子等發揮對造血系統的作用;其“活血”作用主要作用于循環系統,通過抑制血小板凝聚等作用達到“活血”的效果[3]。當歸含揮發油、有機酸、多糖、黃酮等多種化學成分[4-10],其藥理作用廣泛,涉及機體多個系統。當歸含多種有機酸類化合物,其代表為阿魏酸,阿魏酸能抑制血小板凝聚[11],具有抗動脈粥樣硬化作用,阿魏酸對閉塞性動脈粥樣硬化癥也具有顯著療效[12]。當歸中的苯酞類化合物是當歸揮發油的主要成分,包括Z-藁本內酯、E-藁本內酯、洋川芎內酯I、E-丁烯基苯酞、Z-丁烯基苯酞等成分[13]。當歸及其揮發油具有調節血管生成、抑制心肌細胞肥大和抗心律失常作用[14]。
中成藥多指標控制模式近年逐步應用于產品的質量控制,但存在對照品不易獲得、使用量大、檢驗成本高等諸多不足。一測多評法利用藥品各成分之間的內在函數關系,通過對一個對照品易得成分的測定,實現多個成分的含量測定,可大大降低檢驗成本[15-21]。現行當歸丸(濃縮丸)質量標準無含量測定控制項,本研究采用一測多評法同時測定當歸丸(濃縮丸)中的阿魏酸、洋川芎內酯I和藁本內酯3種成分的含量,從而控制本品質量。
1 儀器與材料
1.1 儀器
Waters 2695高效液相色譜儀(美國Waters公司),工作站:Empower;Agilent 1260高效液相色譜儀(美國Agilent公司),工作站:Agilent ChemStation;Mettler Toledo XPE205電子天平(瑞士Mettler Toledo);KQ-00VDE型雙頻數控超聲波清洗器(昆山市超聲儀器有限公司);Milli-Q超純水系統(美國Millipore公司);色譜柱:XSelect?CSH?C18(4.6 mm×250 mm,5 μm),Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)。
1.2 材料
阿魏酸對照品(美國Phenchem公司,批號:KL2510EW,含量:99.09 %);洋川芎內酯I對照品(四川省維克奇生物科技有限公司,批號:WK20032304,含量:98.40 %);藁本內酯對照品(中國食品藥品檢定研究院,含量:100 %,批號:111737-201608);當歸丸(濃縮丸)(仲景宛西制藥股份有限公司,批號:190101,190102,180201,180202,180401,180402)。乙腈、甲酸均為色譜純,水為超純水。
2 方法與結果
2.1 色譜條件
色譜柱:C18(4.6 mm×250 mm,5 μm);流動相:乙腈(A)-0.1 %甲酸(B),梯度洗脫(0~10 min,20 %A→58 %A;10~25 min,58 %A→68 %A;25~33 min,68 %A→68 %A;33~40 min,68 %A→95 %A;40~41 min,95 % A→20 % A;41~50 min,20 %A→20 %A);柱溫30 ℃;檢測波長280 nm;流速1.0 ml/min;進樣量10 μl。
2.2 溶液的制備
2.2.1 對照品溶液的制備 分別精密稱取阿魏酸對照品7.82 mg,洋川芎內酯I對照品 4.34 mg,藁本內酯對照品21.8 mg,分別置入25,25,20 ml量瓶,分別加甲醇配制成阿魏酸,洋川芎內酯I,藁本內酯濃度為309.9,170.8,1090 μg/ml 的儲備液。分別吸取阿魏酸儲備液0.1,0.1,0.1,0.2,0.4,0.6 ml,洋川芎內酯I儲備液0.25,0.25,0.25,0.5,1.0,1.5 ml及藁本內酯儲備液1.0,1.0,1.0,2.0,4.0,6.0 ml,分別置入50,25,10,10,10,10 ml量瓶,加甲醇至刻度,搖勻,配制系列混合對照品溶液。
2.2.2 供試品溶液的制備 取當歸丸(濃縮丸)粉末約1 g,精密稱定,置入150 ml具塞錐形瓶,精密加75 %甲醇50 ml,密塞,稱定重量,超聲處理20 min,放冷,再稱定重量,用75 %甲醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.2.3 陰性對照溶液的制備 本品只含當歸一味藥材,故取制備供試品溶液時所用75 %甲醇溶液作為陰性對照溶液。
2.3 專屬性試驗
取2.2項下3種溶液(其中混合對照品溶液含阿魏酸6.2 μg/ml,洋川芎內酯I 8.5 μg/ml,藁本內酯218.0 μg/ml),分別進樣測定,結果供試品溶液色譜圖中,在與對照品溶液色譜圖相應的位置上,有相同保留時間的色譜峰,阿魏酸、洋川芎內酯I和藁本內酯峰形對稱,與相鄰色譜峰分離度>1.5,理論塔板數按各成分計均≥4000,陰性對照無干擾。見圖1。
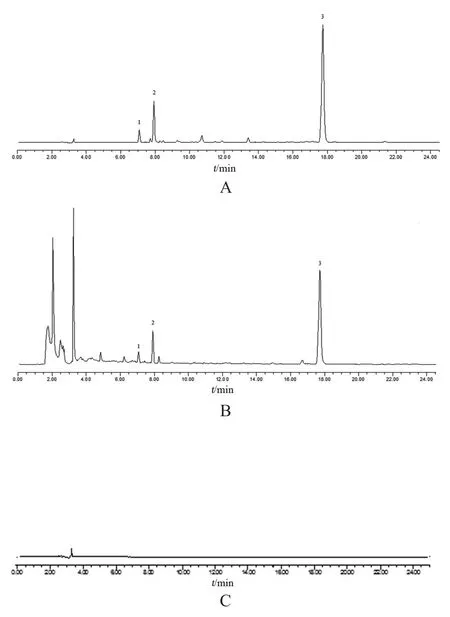
圖1 專屬性考察HPLC圖譜
2.4 線性關系考察
分別精密吸取2.2.1項下系列混合對照品溶液10 μl,注入高效液相色譜儀,按上述色譜條件測定,記錄色譜峰面積,以進樣質量濃度為橫坐標(X),以峰面積為縱坐標(Y),繪制標準曲線,計算回歸方程。結果表明3種成分在相應范圍內與峰面積線性關系良好,見表1。
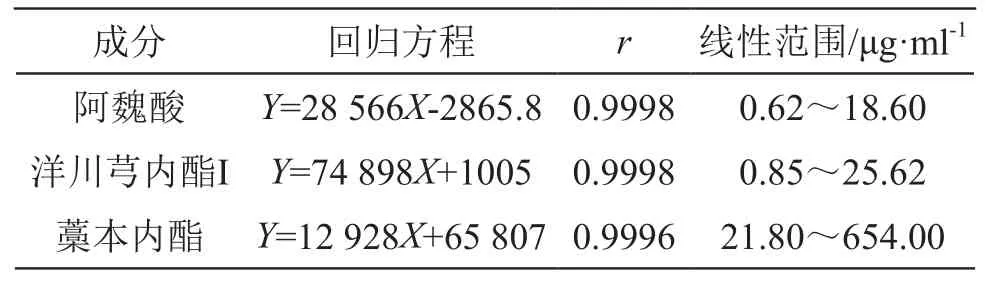
表1 3種成分線性關系
2.5 精密度試驗
精密吸取同一混合對照品溶液(含阿魏酸6.2 μg/ml,洋川芎內酯I 8.5 μg/ml,藁本內酯218.0 μg/ml),連續進樣6次,記錄藁本內酯、洋川芎內酯I、阿魏酸峰面積積分值,計算RSD,結果各成分峰面積的RSD分別為1.11 %,1.28 %和1.18 %,表明儀器精密度良好。
2.6 重復性試驗
取同一批當歸丸(濃縮丸)(批號:190101)粉末約1 g,共6份,精密稱定,分別按2.2.2項下方法制備供試品溶液,在上述色譜條件下分別進樣測定,測得阿魏酸、洋川芎內酯I、藁本內酯含量分別為0.27,0.29,9.12 mg/g,RSD分別為1.61 %,1.56 %,1.91 %。結果表明該方法重復性良好。
2.7 穩定性試驗
取同一批當歸丸(濃縮丸)(批號:190101)供試品溶液,分別于0,2,4,8,12,24 h,按2.1項色譜條件測定,記錄峰面積,結果表明阿魏酸、洋川芎內酯I、藁本內酯峰面積的RSD分別為0.89 %,0.89 %,0.59 %,表明供試品溶液在24 h內穩定性良好。
2.8 加樣回收試驗
取已知含量的供試品(批號:190102)約0.5 g,精密稱定,分別加入一定量的對照品溶液,按2.2.2項下方法制備供試品溶液,按2.1項下色譜條件測定,計算加樣回收率。結果表明當歸丸(濃縮丸)中上述3種成分的平均加樣回收率分別為96.79 %,97.95 %,98.25 %,RSD分別為1.27 %,1.52 %,1.69 %,表明方法準確度良好,見表2。
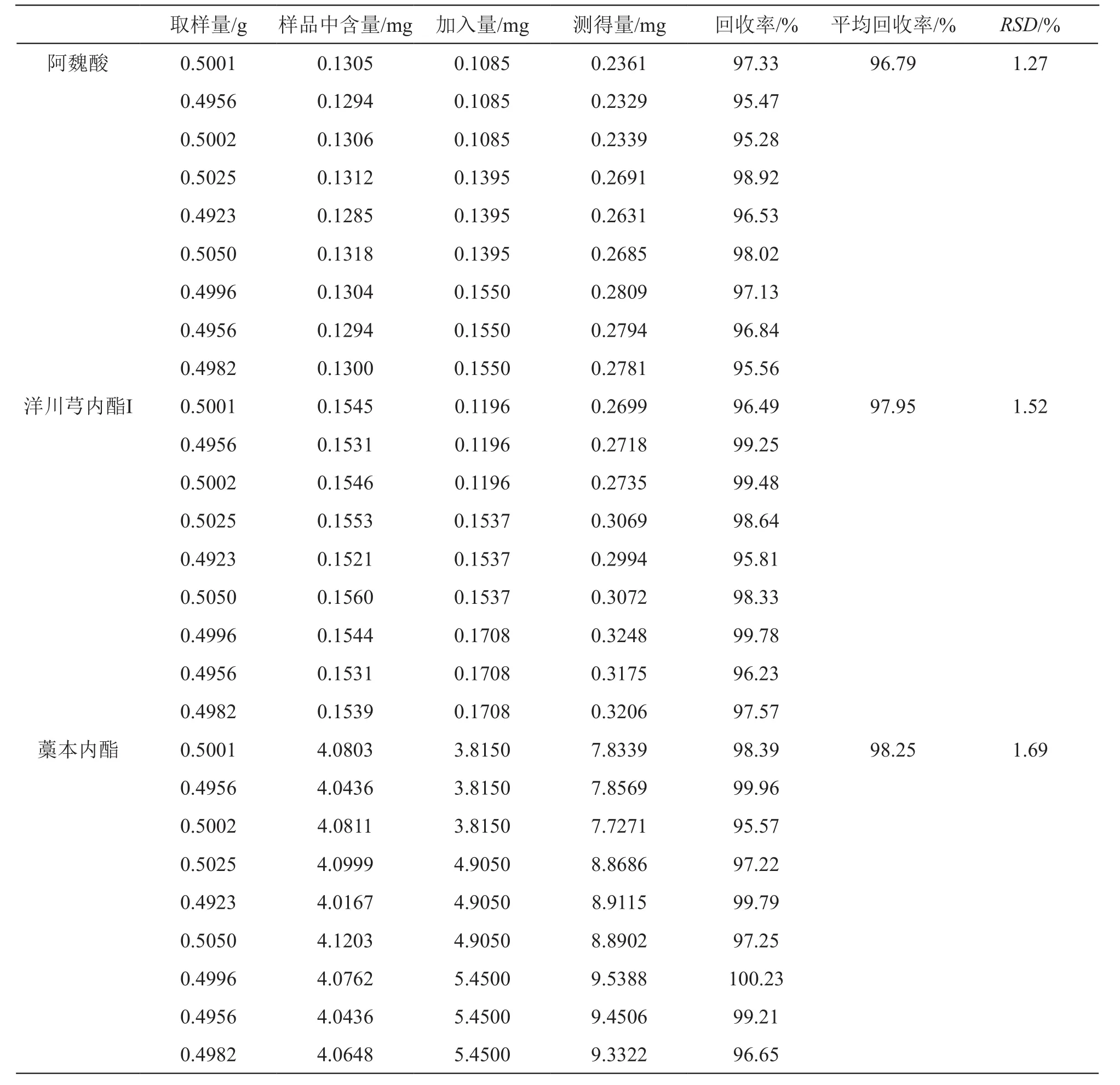
表2 3種成分的加樣回收試驗結果
2.9 相對校正因子的計算
分別精密吸取混合對照品溶液(含阿魏酸6.2 μg/ml,洋川芎內酯I 8.5 μg/ml,藁本內酯218.0 μg/ml)1,2,5,8,10,15,25 μl,注入高效液相色譜儀,按2.1項色譜條件測定,記錄各成分的峰面積,以藁本內酯為內參物,按公式fk/m=fk/fm=(Wk×Am)/(Wm×Ak)(Ak為內參物峰面積,Wk為內參物的質量或濃度,Am為其他成分m的峰面積,Wm為其他成分m的質量或濃度)[22],計算其他成分與內參物的相對校正因子。結果表明,以藁本內酯為內參物,其他成分的fk/m值的RSD均小于5 %,見表3。
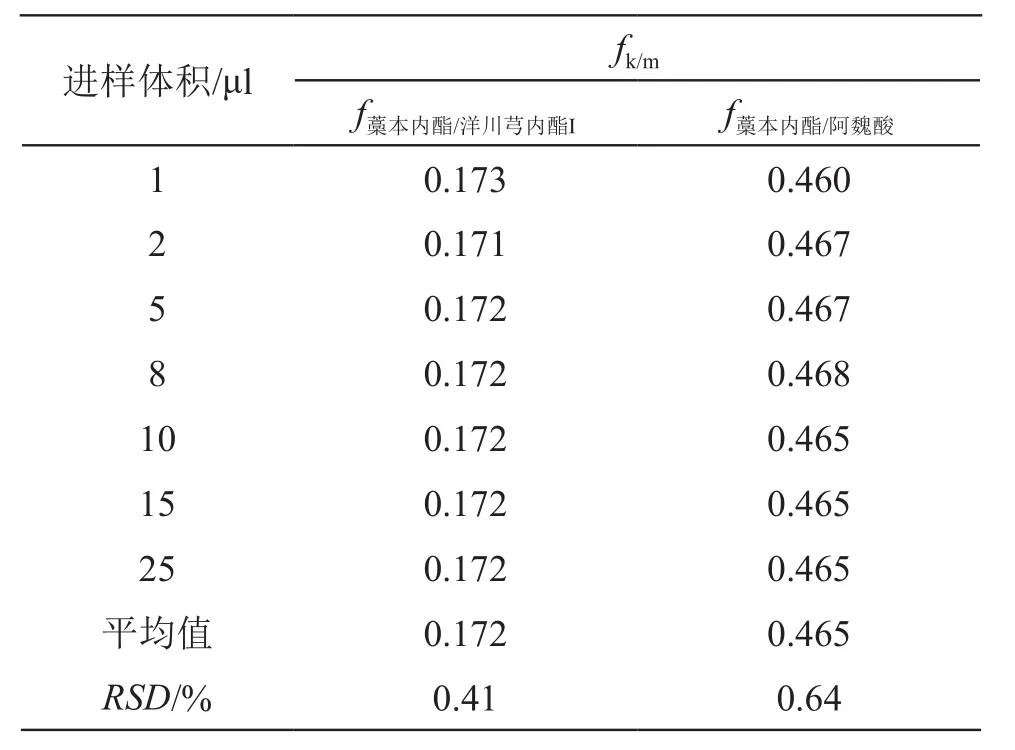
表3 3種成分相對校正因子
2.10 耐用性和系統適應性評價
2.10.1 不同儀器和不同色譜柱考察 取2.2.1項下混合對照品溶液(含阿魏酸6.2 μg/ml,洋川芎內酯I 8.5 μg/ml,藁本內酯218.0 μg/ml),考察Waters2695、Agilent 1260 2種高效液相色譜儀和XSelect?CSH?C18(4.6 mm×250 mm,5 μm)、Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)2種色譜柱對fk/m值的影響,見表4。結果表明,各fk/m值之間無顯著差異,RSD均小于5 %。
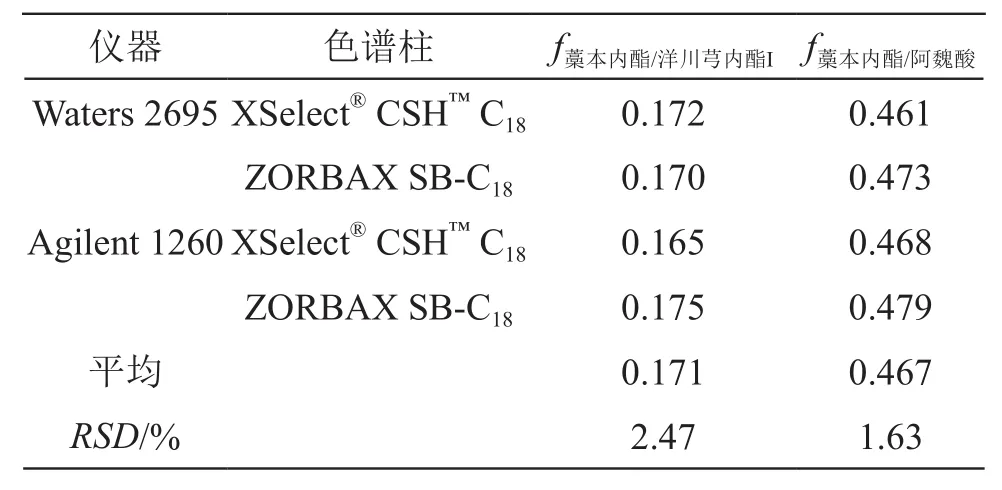
表4 不同儀器、色譜柱對fk/m的影響
2.10.2 不同柱溫的影響 采用Waters 2695高效液相色譜系統,XSelect?CSH?C18柱,考察了柱溫25,30,35,40 ℃時的fk/m,結果表明各成分之間fk/m重復性良好,無顯著差異,RSD均小于5 %,見表5。
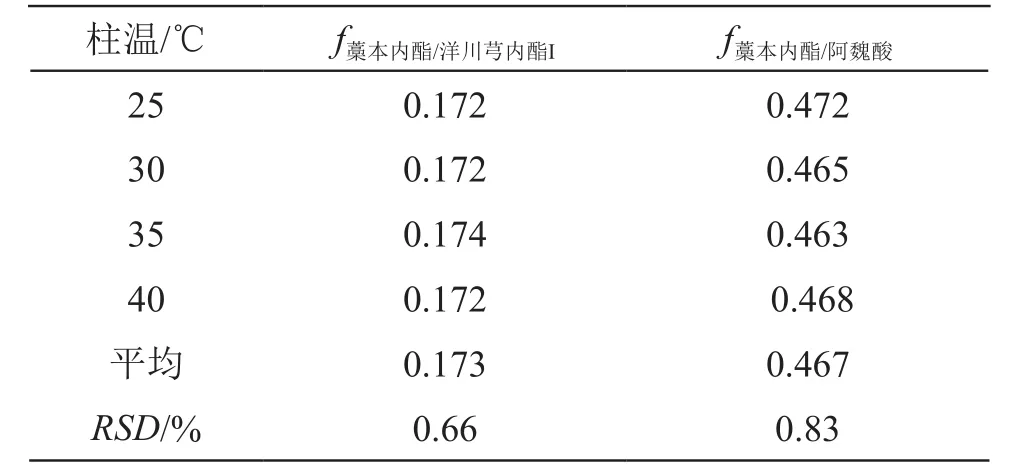
表5 不同柱溫對fk/m的影響
2.10.3 待測組分色譜峰定位 取2.2.1項下混合對照品溶液,采用Waters 2695和Agilent 1260 2種高效液相色譜儀和XSelect?CSH?C18(4.6 mm×250 mm,5 μm)、Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)2種色譜柱,計算待測成分的相對保留時間,對待測組分進行定位[23],并計算RSD。結果各待測成分的相對保留時間的RSD均小于5 %,表明采用相對保留時間對待測成分進行定位是合理的。結果見表6。
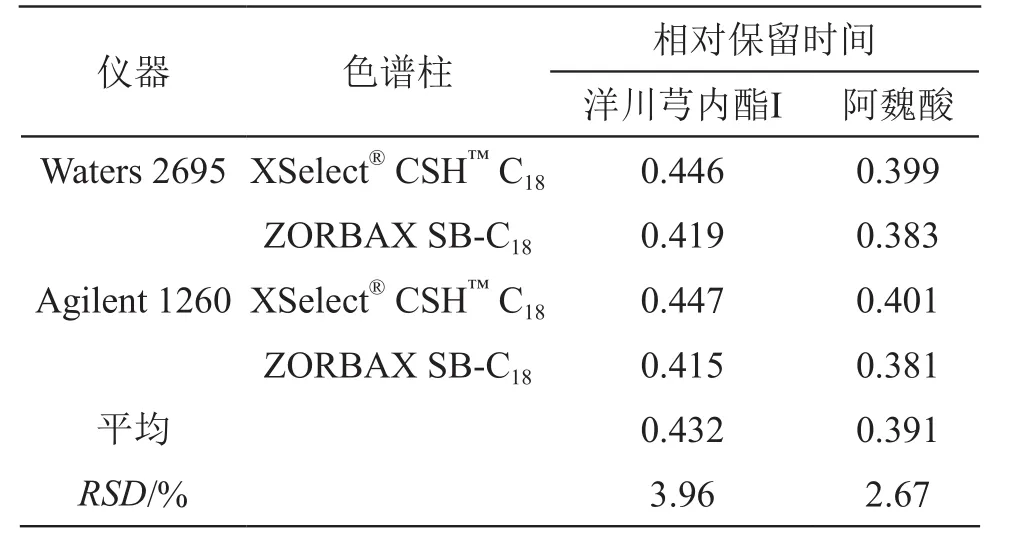
表6 不同儀器和色譜柱對相對保留時間的影響
2.11 一測多評法(QAMS)與外標法(ESM)的數據比較
取6批當歸丸(濃縮丸)樣品,首先采用ESM計算藁本內酯的含量,然后采用ESM和QAMS分別計算樣品中洋川芎內酯I和阿魏酸的含量,并將2種方法的結果進行比較,用相對誤差(RE)來表示2種方法的差異[24],結果見表7。由表7可見,RE絕對值均小于5 %,說明建立的QAMS法有較好的準確性與可行性。
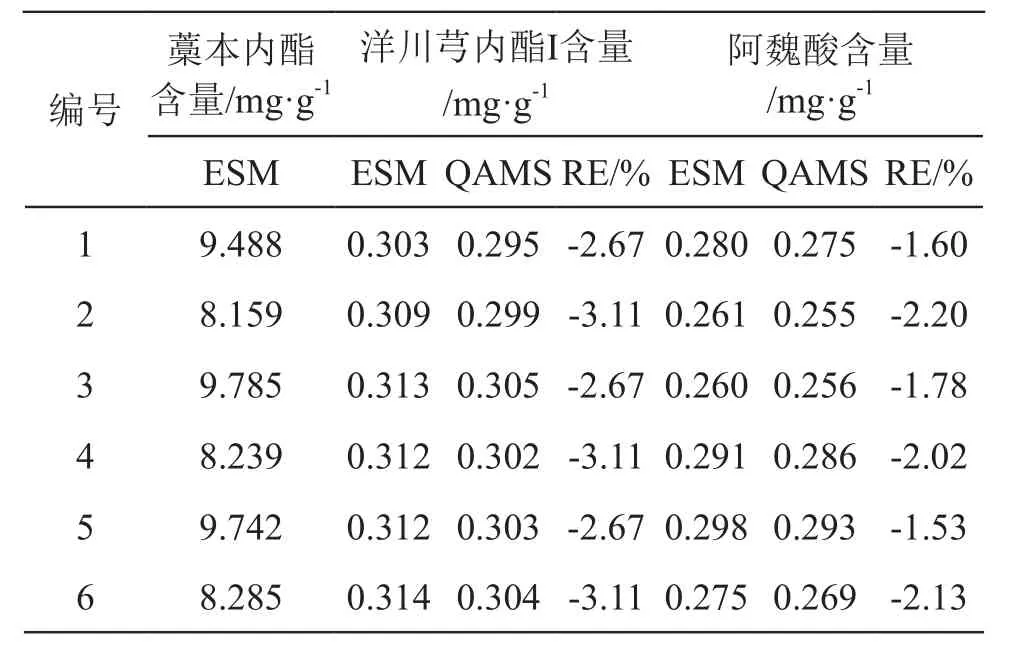
表7 3種成分含量測定結果(n=6)
3 討論
3.1 內參物的選擇
藁本內酯為《中國藥典》2020年版規定的當歸藥材的質量控制指標性成分,性質穩定、市場流通性強、較易得到,且在2.1項下色譜條件下峰面積較大,分離度良好,結合上述實驗數據,證明選擇其作為內參物,建立當歸丸(濃縮丸)的QAMS法可行。
3.2 流動相及柱溫的考察
比較甲醇和乙腈作為有機相、不同濃度的甲酸水溶液和磷酸水溶液作為水相的梯度洗脫效果,結果發現乙腈-0.1 %甲酸水溶液梯度洗脫分離效果較好,達到基線分離,所以選擇乙腈-0.1 %甲酸水溶液梯度洗脫系統為流動相。同時還對4種不同的柱溫:25,30,35,40 ℃進行了考察,根據其對分離效果的影響及對色譜柱壽命的影響,選擇柱溫為30 ℃,此溫度下,各成分分離效果和峰形均較好。
3.3 提取溶劑及提取時間的考察
分別考察了25 %甲醇水溶液、50 %甲醇水溶液、75 %甲醇水溶液、甲醇4種提取溶劑,結果表明75 %甲醇作為提取溶劑時提取效果最好;對超聲提取時間進行考察,分別超聲提取10,20,30,45 min,結果表明,超聲提取20 min時,提取完全。
本研究選用對照品相對易得且化學性質相對穩定的藁本內酯為內參物,建立了當歸丸(濃縮丸)中該成分與洋川芎內酯I和阿魏酸的fk/m。并通過對6批樣品的測定,驗證了當歸丸(濃縮丸)中藁本內酯、洋川芎內酯I和阿魏酸QAMS含量測定的準確性,為當歸丸(濃縮丸)的質量控制提供了參考方法。
