內標—電感耦合等離子體原子發射光譜法在燒結礦中Al2O3、CaO、MgO、MnO和SiO2含量測定中的應用
2022-03-14 10:26:18孫韋青
寶鋼技術 2022年1期
張 艷,沈 健,孫韋青
(寶山鋼鐵股份有限公司制造管理部,上海 201900)
燒結礦是煉鐵主要原料,燒結礦的檢測質量直接關系煉鐵質量,且燒結礦中雜質元素的含量關系到煉鐵及煉鋼的純凈度,所以雜質元素的檢測非常重要。
目前在用的燒結礦中主要雜質元素是Al、Ca、Mg、Mn和Si,對于Al、Ca、Mg、Mn和Si含量檢測的經典化學分析方法中,Si有重量法,Al、Ca、Mg有容量法和原子吸收法,Mn有光度法和原子吸收法等,操作復雜,檢測周期長,過程中使用的試劑量大,很難滿足生產檢驗的需要,所以建立快速、準確的測定方法尤為重要。
目前X熒光光譜儀測定燒結礦中成分因檢測速度快,已成為當今冶金行業生產工藝過程監控的分析趨勢。但X熒光分析方法建立時需要大量的標準樣品建立工作曲線,且日常分析中還需要濕法檢測方法作為比對基礎。
本文建立的內標—電感耦合等離子體原子發射光譜法,適用于不同種類燒結礦中Al2O3、CaO、MgO、MnO和SiO2含量的同時測定。
1 試驗部分
1.1 ICP-AES工作條件
儀器采用VISTA PRO型等離子發射光譜儀(美國Aglient公司)。
發生器功率:1.2 kW;觀察高度:11 mm;載氣流量:0.80 L/min;等離子氣流量:15.00 L/min;輔助氣流量:1.5 L/min;樣品沖洗時間:30 s;積分時間:5 s;積分次數:3次;樣品液的提升量:1.0 mL/min。
1.2 主要試劑
主要試劑為:混合熔劑(無水碳酸鈉+硼酸=2+1)、鹽酸、氧化鐵粉(>99.99%);Al、Ca、Mg、Mn、Si標準儲備溶液(1 000 μg/mL),鈣標準溶液(500 μg/mL),鋁、鎂、錳、硅混合標準溶液(Al 100 μg/mL;Mg 200 μg/mL;Mn 50 μg/mL;Si 250 μg/mL);釔貯備溶液(Y 600 μg/mL)、釔內標溶液(Y 60 μg/mL)。
1.3 試驗內容
1.3.1 試樣的分解
稱取0.500 g(精確到0.000 2 g),加2.0 g混合熔劑于鉑坩堝中,用鉑絲或不銹鋼絲充分混勻;再稱入1.0 g混合熔劑均勻覆蓋在表面,在1 100 ℃的高溫爐中熔融10 min;取出坩堝,待熔融物凝固冷卻,將坩堝置于250 mL低壁燒杯中,燒杯中預先加入70 mL水和20 mL鹽酸,蓋上表面皿,在電爐上加熱,直至熔融物完全溶解,取下燒杯,取出鉑坩堝,用水沖洗干凈,待溶液溫熱后,將溶液轉移入200 mL容量瓶中,用水稀釋到近刻度,待溶液冷卻后,用水稀釋至刻度,混勻。
1.3.2 測定溶液的制備
準確移取溶液(1.3.1)10.00 mL于100 mL容量瓶中,再加入釔內標溶液(60 μg/mL)5.00 mL,鹽酸溶液(1+1)10.00 mL,用水稀釋到刻度,混勻。
1.3.3 測定
按設備操作規程進行儀器操作,設定儀器分析條件,按設備提示,依次噴測工作曲線和試樣待測液。
1.4 工作曲線的繪制
稱取0.40 g基準氧化鐵數份于鉑坩堝中,按照1.3.1隨同樣品進行分解試驗,制備基體溶液,從基體溶液中分取10.00 mL若干份,分別置于100 mL容量瓶中,根據實際需要,添加有濃度梯度的鋁、鎂、鈣、錳、硅標準溶液,再加入釔內標溶液(60 μg/mL)5.00 mL,鹽酸溶液(1+1)10.00 mL,用水稀釋到刻度,混勻。以下按1.3.3操作,測定強度,將測得的強度值與已知含量的對應關系,通過計算機繪制工作曲線。
2 結果與討論
2.1 混合熔劑加入量及熔樣時間
試驗中選擇了鉑金坩堝作為熔融坩堝,使堿熔過程中堝體的引入量極少。針對鉑金坩堝,選擇了適用的熔劑:混合熔劑(無水碳酸鈉+硼酸=2+1)。
為確認熔劑用量和熔融時間,試驗中選擇同一樣品,添加不同的溶劑,采用不同的熔融時間觀察試驗現象,結果見表1。
從檢測結果看,加入不少于2 g熔劑,檢測結果均一致,但從熔樣情況看選擇熔劑用量3 g,熔樣時間10 min。
2.2 熔塊浸取酸量選擇
樣品熔融后,選擇鹽酸溶液浸取。因為在濃酸和加熱條件下,硅易聚合沉淀。為避免硅的聚合沉淀,一則要考慮加熱溫度,二則要考慮鹽酸濃度,試驗選擇加熱溫度保持溶液近沸狀態,鹽酸濃度要盡可能少,所以既要考慮鹽酸的用量,又要考慮鹽酸稀釋用水量,因為考慮熔塊酸化時合適的容積,試驗確認加水量為70 mL。通過試驗確認了鹽酸的浸取熔塊的用量,結果見表2。
從樣品浸取速度和狀態考慮,選擇鹽酸浸取酸量20.0 mL。
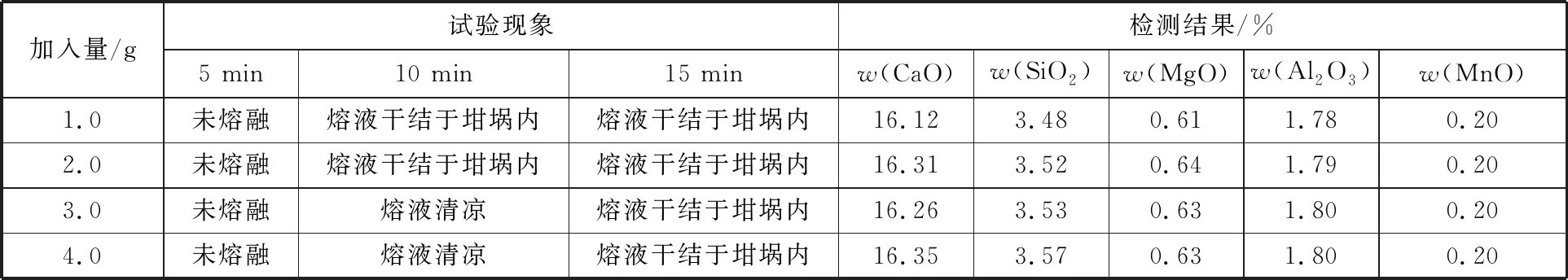
表1 熔劑用量和熔融時間試驗Table 1 Experiment of flux dosage and melting time
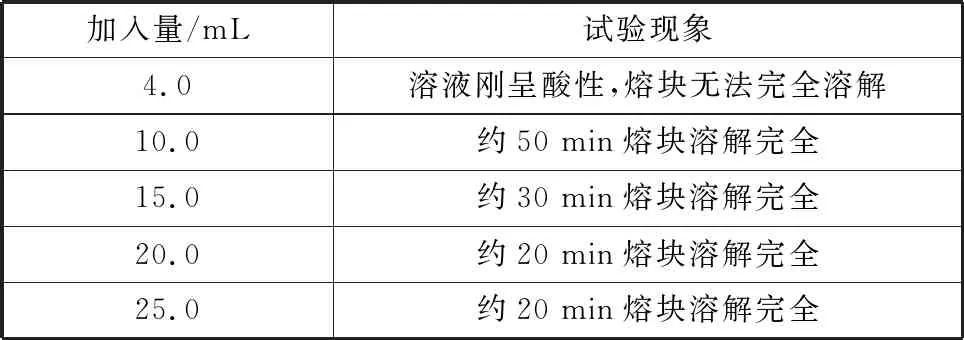
表2 鹽酸浸取用量Table 2 Leaching amount of hydrochloric acid
2.3 分析譜線的選擇
根據儀器軟件譜線庫推薦,依據靈敏度高、元素光譜干擾少、穩定性好的原則,選擇各元素的分析譜線見表3。
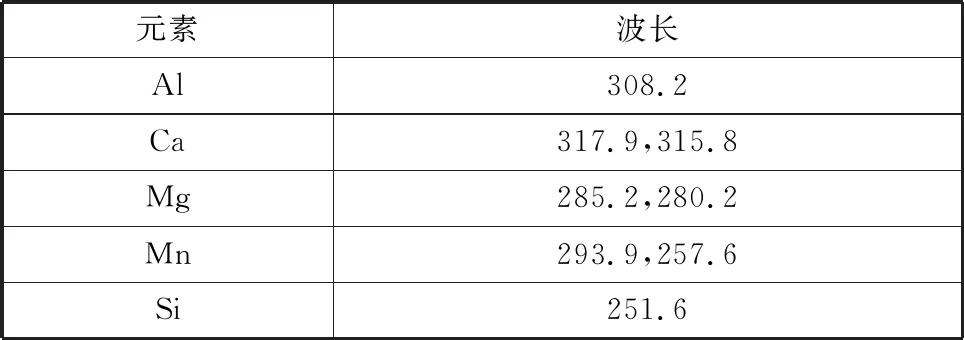
表3 波長分析Table 3 Analysis of wavelength nm
2.4 內標元素選擇
內標元素的選擇首先需考慮在待測樣品中不含或極少含有,且不干擾待測元素;其譜線的發射強度應較大,內標元素測量精度高。另外,在電弧光源和火花光源分析中要求內標元素和分析元素的蒸發速度、電離能及原子量要接近;要求內標線和分析線的激發能和波長接近;要求兩譜線無自吸現象。對于ICP發射光譜分析,由于ICP光源具有高的溫度及獨特的環形結果,提供了很強的蒸發和激發能力。在選擇內標時要求可放低[1]。所以,本方法選擇了ICP內標法常用的釔作為內標元素。
2.5 內標元素加入量選擇
取同一樣品按照試驗方法分解后制備成溶液若干份,分別加入不同量的釔標準溶液,制成待測試液,在給定的儀器工作條件下進行10次測量,得到的相對標準偏差見表4。
電感耦合等離子體原子發射光譜法在低含量段測定本身穩定性較好,內標在電感耦合等離子體原子發射光譜法中的應用,對于高含量段檢測的穩定性提高更加顯著。對于燒結礦中Al2O3、CaO、MgO、MnO、SiO2含量檢測,其中含量較高的成分是CaO,所以試驗中主要考慮內標法對Ca檢測穩定性的提高,通過表4試驗結果,選擇在100 mL試液中加入300 μg的釔內標元素,即60 μg/mL釔標準溶液中分取5.00 mL于100 mL容量瓶中。

表4 不同內標加入量的檢測相對標準偏差Table 4 The relative standard deviation of different internal standard dosage %
2.6 內標元素譜線選擇
內標元素譜線選擇需發射強度大、峰型對稱,且本身測量精度較高。用含釔300 μg/100 mL的試樣溶液在各譜線分別測量10次,其測量精度(相對標準偏差)見表5。

表5 不同內標譜線的檢測相對標準偏差Table 5 The relative standard deviation of different spectral lines of nternal standard %
由表5可見,324.2、361.1、371.0 nm處測量精度優于其他譜線,從譜線的潛在干擾考慮,324.2 nm有Nb、Zr的干擾,361.1 nm有Ce的干擾,所以本方法選擇釔內標分析譜線371.0 nm。
2.7 基體的干擾校正
在燒結礦測定中,主要考慮的基體元素是鐵、鈉和硼的影響。在測定的時候,采取工作曲線溶液中添加與試樣溶液相當量的鐵、鈉和硼,即稱量與試樣相當量的三氧化二鐵和混合熔劑與試樣同時進行熔融浸取,制成基體溶液進行基體匹配,消除基體元素的干擾。同時在檢測時通過背景扣除的方法進一步校正,來補償基體濃度差異所引起的背景漂移。
2.8 共存元素干擾校正
干擾元素的存在將使分析元素產生偏差,干擾校正系數可表征干擾元素對分析元素的干擾程度。為測定共存元素的干擾情況,配制了Al、Ca、Mg、Mn、Si (1 μg/mL)溶液1份,考慮到可能存在的共存元素,同時配制了Si、Ca、Al、Mg、Mn、P、Ti、Na、K、Fe(1 000 μg/mL)10種溶液各1份,分別測定共存元素對分析元素的光譜校正系數[1],得出共存元素對分析元素測定的干擾程度,從試驗干擾情況看,在選定的分析譜線下共存元素的干擾校正系數均很小,共存元素的干擾可忽略不計。
2.9 元素的檢出限和定量限
按試驗方法配制空白溶液,對空白溶液連續測定10次,計算標準偏差,以3倍標準偏差作為方法的檢出限,以檢出限的10倍為方法定量限,結果見表6。

表6 檢出限和定量限 Table 6 Limit detection and limit of quantitation %
2.10 精密度試驗
選擇燒結礦樣品一個,按方法重復試驗11個,并同時設置加內標和非內標模式測定,計算其標準偏差和相對標準偏差(RSD),結果見表7。
從表7結果可見,加內標后檢測結果的精密度均優于不加內標的精密度,方法的穩定性得到明顯提高。

表7 精密度試驗結果(n=11)Table 7 Results of precision tests %
2.11 準確度試驗
本方法采用加標回收試驗來驗證方法的準確性,選擇同一燒結礦樣品加入不同量待測元素,檢測回收量,測定結果見表8。
2.12 不同方法檢測比對試驗
對11個燒結礦,分別采取本方法和國標方法進行檢測,比對結果見表9。

表8 加標回收試驗結果Table 8 Results of recovery tests %

表9 方法比對結果Table 9 Comparison results between two methods %
對檢測數據進行統計分析,得到T檢驗結果tAl2O3=1.37、tCaO=1.48、tMgO=1.43、tMnO=1.84和tSiO2=1.72,都小于臨界值t0.05,10=2.23,說明本方法與國標方法間測定結果是一致的,沒有顯著性差異。
3 結論
試驗表明,內標—電感耦合等離子體原子發射光譜法同時測定燒結礦中Al2O3、CaO、MgO、MnO和SiO2的含量檢測精密度高,相對標準偏差RSD:0.40%~1.52%;準確度好,加標回收率:97%~102%;與國家標準方法間檢測差異性小。且方法檢測速度快,試劑用量小,適用于日常分析測試工作。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
