First principles study on geometric and electronic properties of two-dimensional Nb2CTx MXenes
2022-03-12 07:44:46GuoliangXu徐國亮JingWang王晶XilinZhang張喜林andZongxianYang楊宗獻
Chinese Physics B 2022年3期
Guoliang Xu(徐國亮), Jing Wang(王晶), Xilin Zhang(張喜林), and Zongxian Yang(楊宗獻)
Henan Key Laboratory of Photovoltaic Materials,School of Physics,Henan Normal University,Xinxiang 453000,China
Keywords: Nb2C MXenes,surface functional groups,geometric structure,electronic properties
1. Introduction
Two-dimensional (2D) materials such as graphene,[1,2]transition metal dichalcogenides,[3]phosphorene,[4]etc.have attracted intensive attention and were widely used in energy storage, catalysis, and electronic devices. In recent years, a new family of 2D materials called MXenes[5-7]were routinely synthesized by selectively etching the A-layer elements from the MAX ceramic phases with diluted hydrofluoric acid. The residual 2D MX layers are often covered by surface groups like O,OH,or F,giving a general formula ofMn+1XnTx,whereMis an early transition metal,Xis carbon and/or nitrogen,andTxrepresents surface terminations on the metal layers.Since the first Ti3C2TxMXenes was reported,[5]a large number of MXenes have emerged and shown promising applications in energy storage,[8,9]sensors,[10,11]electromagnetic interference shielding,[12]catalysis,[13]etc.,owing to the charming physiochemical properties. Especially,the intrinsic nature of MXenes vary with elements at the metal site, C/N site, as well as the surface terminations.
For a specific MXenes,surface groups influence the electronic properties and the potential applications because of different electronegativity and stable sites. For instance, the majority-spin and minority-spin electrons of bare Cr2C respectively have metal and insulator properties, giving a halfmetallic ferromagnetic Cr2C, while Cr2CF2and Cr2C(OH)2are antiferromagnetic, because the majority-spin dispersions hold the same characters as the minority-spin electrons.[14]The band dispersions of bare Sc2C showed that the Fermi level is occupied by the Sc-d band,leading to a metallic Sc2C.When it is terminated with F/OH,the Fermi level of Sc2C moves to the center of the energy gap,and Sc2CF2and Sc2C(OH)2become semiconductors. Because O atom needs more electrons than F/OH, and Sc atoms alone cannot provide enough electrons to oxygen group, so parts of O atoms prefer to be adsorbed at the top of C,giving an insulating Sc2CO2.[15]
The above findings demonstrate that both the type and position of functional groups on MXenes surface have a deep influence on the electronic structure of MXenes,and thus the potential applications.Kanet al.[16]used Nb2CT2(T=O/F/OH)MXenes as substrate of Pt/Pd atoms to explore the modification effect of surface functional groups on the stability and activity by density functional theory. Their results showed that the O/F termination has a good fixation effect toward doped Pt/Pd atoms in comparison with the OH groups. In addition, the O/F functional groups can optimize the electronic structure of Pt/Pd atoms, leading to moderate adsorption of oxygenates. Recently, the Nb-based MXenes covered with S, Se, and Te groups were synthesized by combinations of etching and substitution reactions using Lewis acidic/basic molten salts.[17]The etching of Ti3AlC2MAX phase in molten ZnCl2and several other Lewis acidic molten salts above 500°C results in the Ti3C2Cl2MXenes with a pure Cl termination. Similar methods also were used to synthesize other MXenes including Nb2CCl2. Compared with the F/OH functional groups, the halogen functional groups are more likely to participate in a new type of surface exchange reaction to generate S/Se/Te terminated MXenes, due to the weak surface bonding interaction.[17]These MXenes exhibit unique structural and electronic properties, such as a large in-plane lattice expansion and superconductivity. Moreover,Nb2CSe2powder samples were synthesized by the traditional high temperature solid-state synthesis method.[18]The prepared Nb2CSe2samples demonstrated good conductivity,abundant active sites, low cost, and high oxygen evolution activity, because the resultant TMD-MXenes like structures simultaneously have electro-conductive Nb-C layer and rich active marginal Se layer sites.
Although the Nb2C MXenes with different terminations were prepared and investigated for various applications,a fundamental understanding on the effects of surface groups is insufficient. A comparative study on the influence of different functional groups on the geometric configuration, structural stability, electronic nature,etc., is of significance for the further rational design. In this work,the geometries of four functional groups (O, S, Se, and Te) on Nb2C were investigated based on density functional theory. It is found that the most stable sites vary with the functional groups,giving rise to different lattice parameters and orbital hybridization. From the density of states(DOS)and band diagrams,we found that the conductivity of Nb2CTxis affected by both the terminations and their adsorption sites. The electron localization function(ELF) results revealed that the adsorption site ofTxregulate the charger redistribution, changing the interaction strength betweenTxatoms and the Nb2C.The bonding strength of Nb-Txwas analyzed by projected crystal orbital Hamilton population (pCOHP). The results showed that the Nb-Txbonding strength is weakened with the decrease of electronegativity of functional groups(O>S>Se>Te),contributing to the feasible formation of the surface vacancy.
2. Computational methods
The spin-unrestricted calculations were performed within the framework of density functional theory (DFT) using the Viennaab initiosimulation package(VASP).[19]A plane wave cut off energy of 500 eV was used for the plane-wave expansion of the wave function. The potentials at the core region were treated with projector augmented wave (PAW)pseudopotentials.[20]The exchange-correlation energy was represented by a gradient corrected,[20,21]functional proposed by Perdew, Burke, and Ernzerhof (PBE).[22]Brillouin zone integrations were performed using a 9×9×1 Monkhorst-Pack[23]K-point mesh for geometry optimizations, and the convergence threshold was set to be 10-6eV in energy and 0.01 eV/°A in force. The convergence of geometrical parameters was also tested. For density of states calculations the Brillouin zone was sampled using a 13×13×1K-grids. In all the structures, vacuum regions between two adjacent periodic images were over 16 °A to avoid spurious interaction between periodic images. After examining the effects of HubbardUand van der Waals corrections on the geometric parameters of Nb2C(Table S1)and comparing with previous results, we just consider the van der Waals corrections under D3 scheme.[24]The determination of the bonding and antibonding states betweenMandTxatoms was obtained by the projected crystal orbital Hamilton population(pCOHP)[25-28]as employed by the Lobster program. COHP is a partitioning of the band-structure energy in terms of orbital-pair contributions and it is abond-weighted density-of-states between a pair of adjacent atoms. Deringer and coworkers further developed the projected COHP(pCOHP)by re-extracting Hamilton-weighted populations from plane-wave electronicstructure calculations.[26]The IpCOHP stands for the integral value below the Fermi level of pCOHP, it can be understood as the number of bonded electrons shared between two atoms.
3. Results and discussion
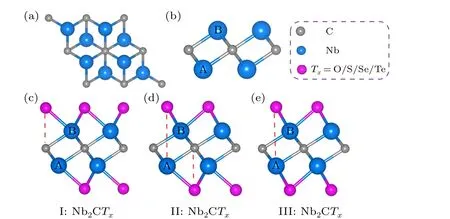
Fig. 1. The structure of Nb2C and Nb2CTx (Tx =O, S, Se, and Te). Top view(a)and side view(b)of 2D Nb2C.Side view of three probable Nb2CTx configurations.
We focused on the geometric configurations and electronic properties of monolayer Nb2C and Nb2CTx(Tx= O,S, Se, and Te). The Nb2C monolayer is composed of three atomic layers stacked in a sequence of Nb(A)-C-Nb(B)(Figs. 1(a) and 1(b)). Three possible configurations of terminations are considered: type I (Fig. 1(c)), the surface groups are located directly above the top site of C atoms;type II (Fig. 1(d)), the groups that are connected to the Nb(A) atoms are at the top sites of the central C atoms and the others are at the hollow sites of Nb(B) atoms, forming an asymmetric arrangement on the Nb2C layer; type III(Fig. 1(e)), the groups are located at the central C hollow sites on both sides of Nb2C layer. In the following section,we will use the nomenclature defined in Fig. 1 for simplification. The optimized Nb2C has an in-plane lattice constant of 3.107 °A accompanied with the Nb-C bond length of 2.15 °A, agreeing with the experimental[29,30]and theoretical reports.[31,32]The total energies of all the MXenes with different termination groups are shown in Table S2 of the supporting information. Upon terminated with O and Te functional groups,configuration III become the most stable structure due to the lowest total energy relative to the other configurations, while the most stable structure is configuration II for the S and Se groups. The phonon dispersions are shown in Fig.S1 to verify the dynamic stabilities. The positive phonon frequencies indicate the dynamic stabilities of III-Nb2CO2,IINb2CS2, and II-Nb2CSe2. The III-Nb2CTe2is dynamically unstable due to the existence of imaginary frequencies. The geometric parameters of Nb2CTx(Tx=O, S, Se, and Te) are summarized in Table 1 for comparatively understanding the influences of functional groups on the structure of MXenes.For the O/S/Se groups,the lattice constant of MXenes gradually increases from configurations I-III as the surface groups migrate. For Te-terminal MXenes,the configuration II has the largest lattice constant. For the same configuration of Nb2CTxMXenes,as the electronegativity of the functional group gradually decreases from O through S and Se to Te, the lattice constant of MXenes and the Nb-Txbond lengths gradually increase,while theTx-Nb-Txangle gradually decreases from O to Te,reflecting the different chemical bond strength between the functional groups and Nb atoms.
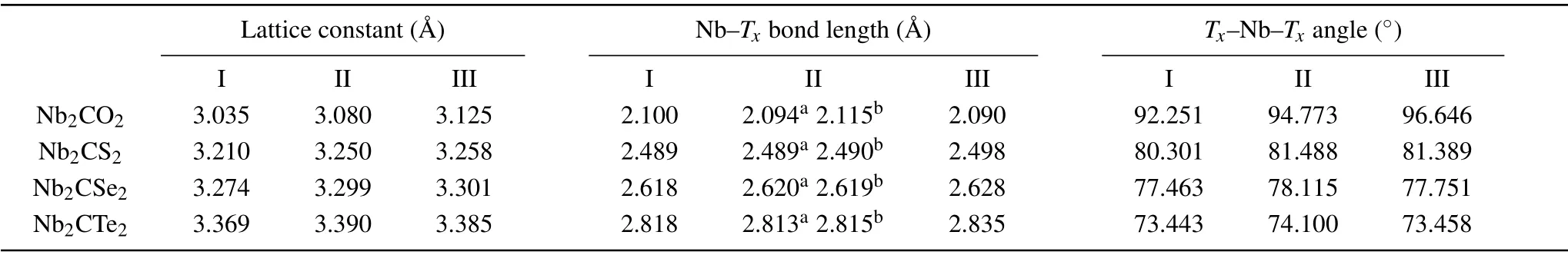
Table 1. The geometric parameters of Nb2CTx (Tx=O,S,Se,and Te)at three configurations. The superscripts a and b mean the bond length of Nb(B)-Tx and Nb(A)-Tx.
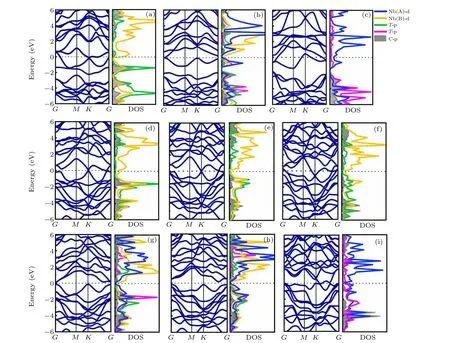
Fig.2.Band dispersions and density of states(DOS)of the Nb2CTx monolayer.(a)I-Nb2CO2;(b)II-Nb2CO2;(c)III-Nb2CO2;(d)I-Nb2CS2;(e)I-Nb2CSe2;(f)I-Nb2CTe2;(g)II-Nb2CS2;(h)II-Nb2CSe2;and(i)III-Nb2CTe2.
The Nb2C MXenes is conductor,since the 4d states of Nb atoms pass through the Fermi level (Fig. S2). The electronic properties of Nb2CTxlayer are strongly related to the functional groups on the surface. When the surface is terminated by O atoms,all the three configurations(Figs.2(a)-2(c))show metallicity. Interestingly,when terminated with S and Se,the resultant MXenes monolayer can be narrow-band gap semiconductors or metals, depending on the arrangement modes.The most stable II-Nb2CS2(Fig. 2(g) and S3(b)) exhibits a semiconducting character with an energy gap of 0.32 eV under the scheme of Heyd-Scuseria-Ernzerhof(HSE06)hybrid functional.[33]The projected DOS analyses show that the conduction band minimum and the valance band maximum are contributed by the d states of Nb(B)and Nb(A)atoms,respectively. The I-Nb2CS2(Fig.2(d))and III-Nb2CS2(Fig.S4(c))are however metallic. Similar to Nb2CS2monolayer,the most favorable II-Nb2CSe2(Fig. 2(h)) has a semiconductor characteristic, while both I-Nb2CSe2(Fig. 2(e)) and III-Nb2CSe2(Fig. S4(f)) are metallic with limited electronic states crossing the Fermi level. When the surface is terminated by Te,the Nb2CTe2(Figs.2(f),2(i),and S4(h))show metal characteristics like Nb2CO2. The above findings indicate that the band structures of MXene are affected by both the types and positions of groups,which are determined by the bonding characters and the orbital hybridization. So the projected densities of states(PDOS)are analyzed to further clarify the contribution of different atoms.
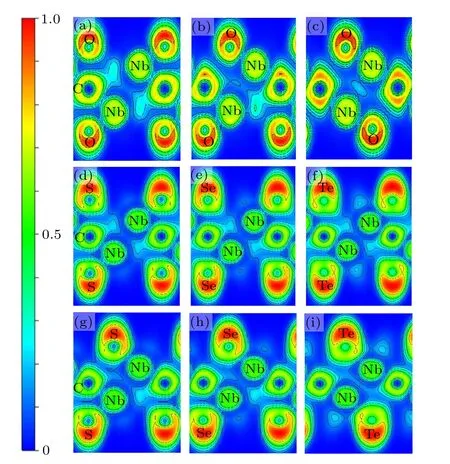
Fig. 3. The 2D slices projected along direction of electron localization function (ELF) are plotted. Upper panel shows the ELF plots for (a) INb2CO2, (b) II-Nb2CO2, (c) III-Nb2CO2, middle panel shows the ELF for(d)I-Nb2CS2,(e)I-Nb2CSe2,(f)I-Nb2CTe2; and lower panel shows(g)IINb2CS2,(h)II-Nb2CSe2,and(i)III-Nb2CTe2.
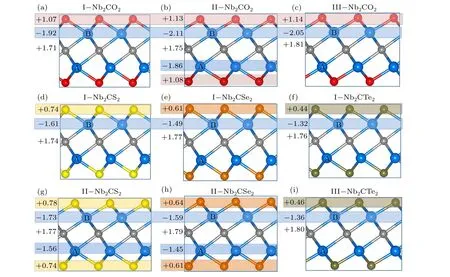
Fig. 4. Bader charge analysis revealing the relative charge transfer between various atoms during the structural phase formation of (a) I-Nb2CO2, (b) IINb2CO2,(c)III-Nb2CO2;middle panel shows(d)I-Nb2CS2,(e)I-Nb2CSe2,(f)I-Nb2CTe2;and lower panel shows(g)II-Nb2CS2,(h)II-Nb2CSe2,and(i)III-Nb2CTe2. Plus symbol“+”indicates the gain of electrons and minus symbol“-”indicates the loss of electrons.
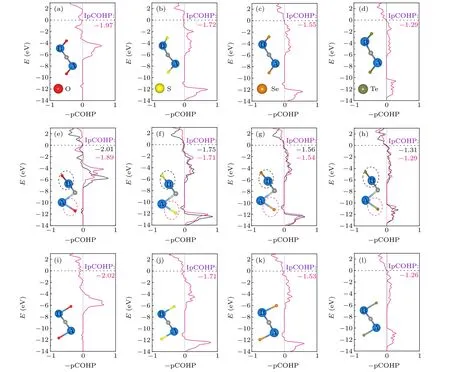
Fig.5. The projected crystal orbital Hamilton population(pCOHP)states have been plotted for the Nb2CTx. (a)-(d)I-Nb2CTx (in which Tx represents O,S,Se,and Te);(e)-(h)II-Nb2CTx (in which Tx represents O,S,Se,and Te);(i)-(l)III-Nb2CTx (in which Tx represents O,S,Se,and Te).
As shown in Fig. 2(a), at the configuration I, the weak interactions among Nb, O, and C atoms are supported by the negligible overlap between Nb-d states with O/C-p states.For the configuration II (Fig. 2(b)), a strong overlap between Nb(B)-d and C/O-p in the energy range of-1 eV to 0 eV and-4.5 eV to-3 eV makes the structure more stable. In configuration III (Fig. 2(c)), an even strong overlap between Nb-d and O/C-p can be observed from-6 eV to Fermi level,indicating a strong bonding interaction between these atoms.Different from Nb2CO2,the configuration II of Nb2CS2has a strong hybridization between Nb(B)-d and C/S-p in the energy range of-3.5 eV to-1 eV and-6 eV to-4.5 eV,making it stable. The similar phenomena are observed on the Nb2CSe2and Nb2CTe2.
In order to further study the effect of surface functional groups on the structural stability,we performed an electron localization function(ELF)analysis(Fig.3). The value of ELF between two atoms can be in the range of 0 to 1, where 1,0.5,and 0 represent covalent,metallic,and no-bonding characters, respectively.[34,35]Taking Nb2CO2as examples, a weak electron-gas between Nb and C together with a highly concentrated electrons around C are observed,showing the metallicionic character of the Nb-C bonds.This conclusion is applicable to Nb-O bonds. For configuration II(Fig.3(b)),when one O atom is migrated to the hollow site, an increased localized electron around O enhances the ionic bond between Nb and O, and this is conducive to the improved stability. In configuration III(Fig.3(c)),when all O groups locate at the hollow sites, the electrons localized around C and O increase obviously,which makes it more stable. These results may explain why the O groups prefer to the hollow site.
When the S atom is placed at the hollow site of Nb(A)atoms (configuration II and Fig. 3(g)), the localized electrons among Nb, C, and S are larger than the configuration I and configuration III (Figs. 3(d) and S6(c)), demonstrating the strong bonding interaction and structural stability.Nb2CSe2has similar bonding characters to Nb2CS2,as shown in Figs.3(e)and 3(h). The ionic characters of Nb-C and Nb-Se can be concluded from the large ELF value of near the C and Se atoms in configuration II.In addition,in configuration III of Nb2CTe2, Nb atoms form a strong ionic bond with the surface Te atoms.
We further performed the Bader charge analyses in Fig.4 to quantitatively estimate the adsorption strength of surface groups on Nb2C surface. It is found that the charge is always transferred from Nb to C and surface groups. In comparison with configurations I and II,the O and C atoms in configuration III gain more electrons, therefore enhancing the bonding interaction between Nb and O/C atoms. It also explains why the O atoms tend to adsorb at the hollow sites.From Figs.4(d)and 4(g), the Nb2CS2in configuration II can transfer more electrons from Nb(B)atom to C and S atoms,while the electron transfer from Nb(A)is fewer,in comparison with that in configuration I. The similar situation is found for the surface terminated by Se and Te functional groups.
To further assess the bond strength of Nb2CTxMXenes,the projected crystal orbital Hamilton population (pCOHP)calculations were carried out. Figure 5 shows the integrated pCOHP dispersions and the corresponding integration values,which are efficient measure of the bonding strength. Compared with the Nb-C bonding contribution,Nb-Txbonding is a dominant factor for surface bonding strength. With the decrease of electronegativity of functional groups, the bonding strength of Nb-Txgradually decreases for the same configuration. The structure with a weak Nb-Txchemical bond strength is easier to form surface vacancy. Obviously, compared with other structures, the IpCOHP value of Nb-Te in configuration III (Fig. 5(l)) is the smallest, demonstrating the weakest bonding interaction and the easiest formation of surface vacancy, in line with the experimental element analyses.[18]For configuration II, the distinct adsorption sites for surface groups lead to imbalanced interaction strength associated with different IpCOHP value(Figs.5(e)-5(h)),the bond strength of Nb(B)-Txis generally stronger than that of Nb(A)-Tx.
4. Conclusion
In conclusion, based on density functional theory calculations, we systematically studied the structural stability and electronic properties of the 2D Nb2CTx(Tx=O, S, Se, and Te),with particular attention on the effects of surface groups.It is found that the O and Te groups prefer to the central C hollow sites on both sides of Nb2C,while the most stable sites for the S and Se terminations are unequable on the two Nb2C sides. The interaction strength between Nb atoms and functional groups decrease gradually from O through S and Se to Te. The bonding interaction was analyzed by the electron location function, Bader charge, and the projected crystal orbital Hamilton population. From O to Te groups, the surface terminations gain fewer electrons, and the integrated pCOHP value gradually reduced. The changed interaction will modify the electronic nature of Nb2C MXenes. The types of surface groups dominate the electronic characters and conductivity, and the adsorption sites are the secondary influencing factor. The Nb2CO2and Nb2CTe2always have metallicity.The Nb2CS2and Nb2CSe2can be semiconductor or metal,depending strongly on the adsorption sites of surface groups on Nb2C.We anticipate that the present results could provide some insights into the fundamental understanding toward the effects of surface groups to the MXenes.
Acknowledgments
Project supported by the National Natural Science Foundation of China (Grant Nos. U1804130, U2004212,11904084, and 11874141), the Henan Overseas Expertise Introduction Center for Discipline Innovation (Grant No.CXJD2019005),the China Postdoctoral Science Foundation (Grant No. 2021M690933), and the Key Scientific Research Projects of Henan Education Department,China(Grant No. 22A140020). The simulations are performed on resources provided by the High Performance Computing Center of Henan Normal University.

- Chinese Physics B的其它文章
- Surface modulation of halide perovskite films for efficient and stable solar cells
- Graphene-based heterojunction for enhanced photodetectors
- Lithium ion batteries cathode material: V2O5
- A review on 3d transition metal dilute magnetic REIn3 intermetallic compounds
- Charge transfer modification of inverted planar perovskite solar cells by NiOx/Sr:NiOx bilayer hole transport layer
- A low-cost invasive microwave ablation antenna with a directional heating pattern