誘導自噬對朊病毒病治療、作用機制的研究進展
2022-03-05 07:55:04黃雨妍李睿涵李佳津韋文君緱夢蘭汪寧
系統醫學 2022年22期
黃雨妍,李睿涵,李佳津,韋文君,緱夢蘭,汪寧
1.中國醫科大學第一臨床學院,遼寧沈陽 110001;2.中國醫科大學法醫學院,遼寧沈陽 110122
朊病毒病是由于異常折疊的朊蛋白——朊病毒蛋白(prion protein,PrPSc)的大量聚集引起的神經退行性疾病。而自噬作為體內清除錯誤折疊的蛋白質或抑制聚集的主要方式,通過多種途徑如轉錄因子EB(transcription factor EB,TFEB)調控的自噬溶酶體途徑(autophagy-lysosome pathway,ALP)、哺乳動物雷帕霉素靶蛋白(mammalian target of rapamy‐cin,mTOR)介導的自噬抑制途徑和AMP 活化激酶(adenosine 5’-monophosphate-activated protein ki‐nase,AMPK)介導的ALP 途徑等參與朊病毒病的發生,并成為在研究疾病的進展與治療中不可忽視的存在,得到了廣泛的研究。自噬在朊病毒病中的作用及作用機制并不明確,此外,過度自噬會加重朊病毒病的進展,這一觀點引起了科學界的廣泛關注。近年來,越來越多的科研工作者針對自噬在朊病毒病中的作用機制進行了大量研究,研究表明許多基于誘導自噬的藥物在小鼠試驗階段甚至臨床試驗階段都呈現出良好的治療效果。文章總結了自噬在朊病毒病中的作用機制,詳細闡明了誘導自噬對朊病毒病所產生的積極影響,這對探索朊病毒病的治療具有重要意義。本文對臨床常用藥物如雷帕霉素、甲磺酸伊馬替尼、白藜蘆醇等進行了簡單介紹,并簡述了相關作用機制。現報道如下。
1 朊病毒病
朊病毒病也稱為傳染性海綿狀腦病(transmis‐sible spongiform encephalopathy,TES),是一種能夠感染人類和動物的致命性神經退行性疾病[1]。人類朊病毒病根據發病形式可以分為:①散發性人類朊病毒疾病,包括克魯茨費爾特-雅各布病(creutzfeldtjakob disease,CJD)、致死性失眠和可變蛋白酶敏感性朊病毒病;②遺傳性或家族性朊病毒病,一般是由相關編碼基因突變所引起,包括家族性或遺傳性克雅氏病、致命性家族性失眠癥和格斯特曼綜合征(gerstmann-str?ussler-scheinker,GSS);③獲得性人類朊病毒病,包括庫魯病、醫源性克雅氏病和一種新的克雅氏病變異形式,僅占人類朊病毒病病例的5%[2]。在朊病毒病中,人體內與朊病毒病相關的正常蛋白(PrPC)的構象發生轉變,形成一種錯誤折疊、富含β-折疊且易于聚集的病理形式,可稱為朊病毒蛋白(PrPSc)[3]。在未感染的細胞中,PrPC通過內吞途徑降解,然而對比PrPC易于被溶酶體水解酶降解,PrPSc對蛋白水解更具抵抗性,PrPSc的聚集能有效阻止朊病毒蛋白的降解[4]。體內錯誤折疊的毒性蛋白大量聚集[5],通過細胞凋亡使神經元丟失[6],造成嚴重的神經退行性變。
2 自噬
自噬指將細胞質貨物輸送到溶酶體的細胞降解途徑,即細胞“吃掉自己”的過程,其不僅參與胞內物質運輸,還起到清除細胞內受損的細胞器或錯誤折疊的蛋白質等廢物并循環利用相關降解產物的作用,對維持人體正常的生理功能以及內環境的穩態至關重要。自噬通常根據其特定的生理功能、物質運輸方式和途徑分為3 類,分別為巨自噬、微自噬和分子伴侶介導自噬[7]。其中巨自噬最為普遍與典型,在此過程中,細胞接受自噬誘導信號后產生具有雙層膜的吞噬泡,吞噬泡包裹受損的細胞器和蛋白形成自噬體,自噬體內的物質被轉運到溶酶體并在酶的作用下進行降解,產生的大分子被釋放回細胞質中以供再利用[8]。
3 自噬針對朊病毒蛋白的作用機制
在朊病毒病中,自噬具有降解聚集蛋白、減少聚集蛋白數量和降低其毒性的作用。上調自噬可以有效防止神經變性,從而減緩疾病的發展。ALP降解是細胞清除異常蛋白的主要方式。ALP 的缺陷會導致蛋白質聚集、有毒蛋白種類的產生和積累以及細胞器的功能失調。ALP 途徑的作用機制主要概括為TFEB 調控的ALP 途徑、mTOR 介導的自噬抑制途徑、AMPK 介導的ALP 途徑,這3 個途徑既相互獨立,又彼此聯系。
3.1 TFEB 調控的ALP 途徑
TFEB 是一種與溶酶體調控基因或協調溶酶體表達和調控(coordinated lysosomal expression and regulation,CLEAR)元件表達的啟動子結合的轉錄因子[9],通過驅動自噬和溶酶體基因的轉錄,來促進自噬體溶酶體的形成[10]。進一步研究發現,TFEB增強了溶酶體蛋白的穩態,這些蛋白大多是發生了突變或錯誤折疊,同時TFEB 可以促進自噬體與溶酶體的融合[11]。TFEB 的亞細胞定位和活性受磷酸化水平的控制[12],激活后的TFEB 可迅速由細胞質轉位到細胞核與靶序列基因結合,參與自噬的轉錄調控,誘導自噬[10]。在營養豐富的條件下mTOR 復合物1(mammalian target of rapamycin 1,mTORC1)可通過PI3K-Akt 途徑被激活并使TFEB 磷酸化,磷酸化的TFEB 與14-3-3 蛋白結合,留在胞質,無法進入細胞核而失去驅動功能。壓力條件下,mTORC1通過AMPK 途徑失活,去磷酸化的TFEB 發生質核轉位[13],發揮轉錄因子的功能。此外,Ca2+依賴的鈣調神經磷酸酶的激活也可使TFEB 去磷酸化進入細胞核(圖1)[14]。最新研究表明,microRNA(miRNA)可以控制TFEB 的表達和激活,調節TFEB 誘導的CLEAR 基因的轉錄[15]。

圖1 TFEB 的激活途徑
3.2 mTOR 介導的自噬抑制途徑
mTOR 是一種絲氨酸/蘇氨酸激酶,是哺乳動物自噬中研究最好的自噬調節因子,也是mTOR 復合物的催化亞基。根據結構差異,可以將mTOR 復合物分為mTORC1 和mTORC2[16]。
mTORC1 是營養和生長因子信號的主要傳感器,可以影響生長因子、營養物質和細胞能量狀態等多種輸入信號。mTORC1 由富含脯氨酸的40 kDa的Akt 底 物(PRAS40)、mTOR 相互作 用蛋白(DEPTOR)、mTOR 的調節相關蛋白(Raptor)、G 蛋白β-亞基樣蛋白(GβL)和mTOR 構成。當營養豐富的條件下,Akt 作用于PRAS40,使磷酸化的PRAS40 從Raptor 上解離后激活ULK1,抑制自噬溶酶體的形成。
mTORC2 由GβL、mTOR、mTOR 的雷帕霉素不敏感伴侶(Rictor)、應激激活蛋白激酶相互作用蛋白(SIN)1 和與Rictor 觀察到的蛋白(protein ob‐served with Rictor,PROTOR)構成。饑餓條件下,mTORC2 通過FoxO3 信號通路調控自噬。mTORC2磷酸化作用于Akt,Akt 使FoxO3 基因表達的蛋白質(FoxO3)磷酸化,后者與14-3-3 蛋白結合,留在胞質,無法進入細胞核,抑制自噬基因轉錄的激活(圖2)[17]。最新研究表明,mTORC2 可能也存在其他介導途徑,多種途徑共同負調控自噬基因的轉錄。
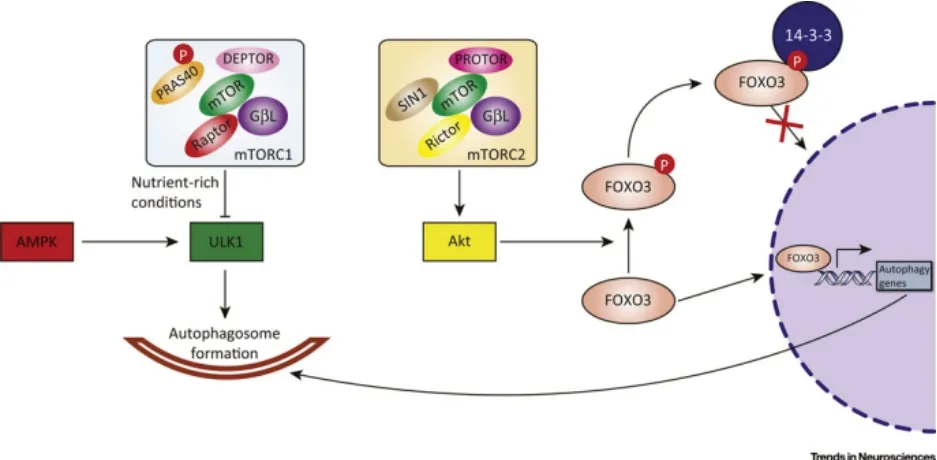
圖2 mTOR 介導的自噬抑制途徑
3.3 AMPK 介導的ALP 途 徑
AMPK 可以響應能量狀態與細胞質鈣水平來進行自噬的調節。在營養充足時,三磷酸腺苷(ad‐enosine triphosphate,ATP)結合AMPK 的γ 亞基使其失活;在饑餓狀態下,一磷酸腺苷(adenosine mono‐phosphate,AMP)結合于AMPK 的γ 亞基,通過腫瘤抑制激 酶(liver kinase B1,LKB1)使α 亞基上 的Thr172 磷酸化,激活AMPK,激活后的AMPK 可通過磷酸化mTORC1 上的Raptor 與TSC1/2 復合物使mTORC1 失活,阻斷mTOR 介導的自噬抑制途徑[18],達到誘導自噬的作用(圖3)[19]。
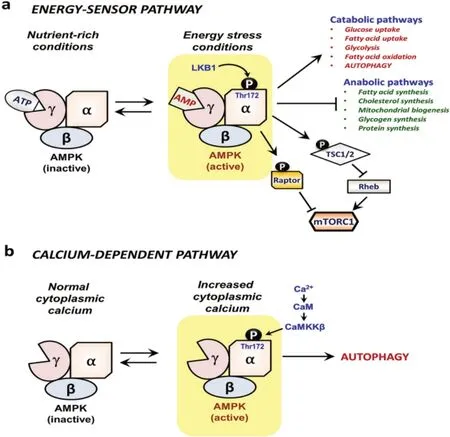
圖3 AMPK 激活的兩種途徑
AMPK 還可以以AMP 獨立的方式被激活,主要與細胞質中鈣的含量有關。神經退行性疾病模型中反復觀察到胞質Ca2+水平異常升高[20]。究其原因為鈣蛋白酶介導的溶酶體膜完整性被破壞,從而導致溶酶體中大量組織蛋白酶泄漏,鈣蛋白酶在細胞內結構中的致病性激活導致大腦中廣泛的退行性疾病[21],特別是散發性克雅氏病,這是與朊病毒疾病相關的神經毒性的關鍵決定因素之一。細胞質鈣是AMPK 和通過鈣調蛋白/CaMKKβ 途徑自噬的激活劑。細胞內鈣水平的增加可使鈣/鈣調蛋白依賴性蛋白激酶激酶2(recombinant calcium/calmodu‐lin dependent protein kinase kinase 2,CAMKK2)被激活,激活后的CaMKK 以AMP 獨立的方式磷酸化α亞基上Thr172,激活AMPK(圖4)[15]。AMPK 激活后,可使自噬通量大大增加。AMPK 誘導的自噬與TFEB 轉錄因子的核轉位有關。

圖4 AMPK 介導的自噬途徑
保持鈣的穩態和鈣介導的AMPK 信號傳導、糾正不平衡的鈣蛋白酶-組織蛋白酶激活,可以更好地確保自噬功能的正常發揮,同時減輕鈣超載帶來的線粒體損傷等其他病理變化[22]。
4 利用自噬通路治療朊蛋白病的藥理學及病理學理論基礎
4.1 自噬的藥理誘導可減少朊病毒蛋白并減輕朊病毒病
各種體內、體外研究表明,自噬誘導會干擾朊病毒感染[23]。在持續性朊病毒感染的細胞或小鼠中研究了幾種自噬的藥理學誘導劑。其中一些化合物通過抑制mTOR 誘導自噬,對自噬進行負調節,其他化合物則在mTOR 非依賴性途徑中工作[24]。
4.2 p62-Keap1-NRF2-ARE 信號通路
主動自噬可以從兩個方面對神經元起到保護作用,降低氧化應激水平。一方面通過減少錯誤折疊的朊病毒數量,另一方面p62-Keap1-NRF2-ARE信號通路中NRF2 由磷酸化p62 觸發從Keap1 上脫離,促使NRF2 轉移到細胞核并激活抗氧化防御機制[25]。
通過p62-Keap1-NRF2-ARE 信號通路誘導的自噬可能為治療常見神經退行性疾病提供有顯著效果的新選擇。由于藥理學NRF2 激活對疾病的廣泛作用,作者推斷絕大多數神經退行性疾病都將適合以此途徑進行靶向治療。
5 各藥物對阮蛋白病的臨床調節作用
朊蛋白病是一類具有傳染性的朊蛋白導致的散發性中樞神經系統變性疾病。目前的醫療水平對朊病毒尚無有效的對因治療辦法,主要是進行對癥的治療。病程遷延數年者很少見,在治療朊蛋白病過程中較常使用的藥物有如下幾種。
5.1 甲磺酸伊馬替尼(Gleevec,STI571)
甲磺酸伊馬替尼(Gleevec,STI571)是一種酪氨酸激酶c-Abl 的抑制劑[26]。該藥處于小鼠實驗階段,感染早期可明顯降低PrPSc在小鼠脾臟中的負荷,主要在中樞神經系統外發揮抗朊病毒蛋白的作用,從而延長小鼠的存活時間。對于嚴重感染后期,該藥物是否能持續發揮顯著效果,以及如何確定最佳的給藥量仍然是需要探究的問題。
5.2 AR-12(OSU-03012)
AR-12 屬于塞來昔布的一種衍生物,也被稱為OSU-03012,具有強大的抗朊病毒作用[27]。該藥處于細胞試驗階段,治療2 周后,AR-12 在持續感染朊病毒的神經元(ScN2a)和非神經元(ScMEF)培養細胞中完全消除了朊病毒感染,然而AR-12 誘導自噬的具體機制尚不明確。
5.3 白藜蘆醇(3-4'-5-trihydroxystilbene)
葡萄中富含的天然酚和植物毒素可以治愈SMBs15 細胞中的朊病毒感染[28]。白藜蘆醇的抗朊病毒和神經保護作用主要歸因于通過激活Sirt1(Ⅲ類組蛋白去乙酰化酶)誘導自噬[29]。
5.4 其他藥物
PPS、奎納克林通過穩定PrPC并減少其向PrPSc的轉化來防止PrPSc聚合物的積累。姜黃素(Cur)具有抗淀粉樣蛋白特性和神經保護作用,親脂性Cur可穿過血腦屏障在腦中抑制朊病毒蛋白的聚集[30]。其他的有抗朊病毒作用的藥物有剛果紅、馬來酸氟吡啶、硫磺素、鐵的四吡咯衍生物。
5.5 典型藥物的藥理機制與作用效果
5.5.1 自噬誘導劑雷帕霉素(Rapamycin)雷帕霉素是經典的哺乳動物雷帕霉素靶蛋白(mTOR)抑制劑和被認可的自噬誘導物,可延長小鼠存活時間。相較于甲磺酸伊馬替尼,雷帕霉素可順利通過血腦屏障,在中樞神經系統發揮作用。已經確定自噬誘導劑雷帕霉素可應用于臨床治療,可以通過靶向抑制mTOR 依賴的自噬抑制途徑來降低PrPSc的聚集水平。
雷帕霉素的靶標(target of rapamycin,TOR)是一種進化保守的絲氨酸/蘇氨酸激酶,可感知和整合來自環境的信號,以協調發育和代謝過程[31]。TOR在條件有利時感知營養、激素、代謝物和壓力信號,以促進細胞和器官的生長。然而,當條件不利時,TOR 會受到抑制,從而促進自噬等分解代謝過程。
5.5.2 候選藥物二甲雙胍(Metformin)有研究表明,二甲雙胍對神經退行性疾病有治療作用。二甲雙胍減少了朊病毒感染,并誘導持續感染22 L 朊病毒的N2a 細胞自噬。用二甲雙胍處理22 L-ScN2a 細胞7 d 可降低朊病毒載量。誘導自噬可能是二甲雙胍減輕N2a 細胞中朊病毒感染的機制之一。
此外,二甲雙胍誘導持續感染RML 朊病毒的ScCAD5 細胞自噬。朊病毒由具有不同構象穩定性和生化性質的各種菌株組成。采用二甲雙胍治療RML 感染的ScCAD5 細胞,10 d 后可減少朊病毒感染,這表明二甲雙胍的體外抗朊病毒作用可能與菌株和細胞類型無關,但抗病毒作用的效力在不同的菌株與細胞類型之間有差異。
為測試二甲雙胍在體內的治療效果,接種并使小鼠在腦脊髓內感染22 L 朊病毒。從接種之日起,在飲用水(2 mg/mL)中用二甲雙胍對小鼠進行治療110 d。與對照組相比,結果顯示二甲雙胍治療組的存活時間沒有增加,并且接受二甲雙胍的小鼠沒有表現出任何低血糖現象。多項實驗表明,二甲雙胍只能暫時降低朊病毒負荷,并不能根除朊病毒感染,長期使用無法治愈感染細胞。短期使用對癥狀有改善作用,長期使用對疾病轉歸的影響有待深入研究。
以上結果表明,自噬在朊病毒病中起到至關重要的作用,在患者和實驗接種小鼠的大腦中也可以觀察到自噬體。然而,這種水平的自噬不足以解決朊病毒感染。使用化合物增強自噬已被證明可以增加PrPSc降解,限制其外泌體釋放,并具有神經保護作用。雖然這些通過自噬干擾朊病毒繁殖的多種方式在體外是高效的,但只有少數化合物可以有效地提高朊病毒感染小鼠的存活率、延長存活時間,臨床試驗階段仍處于萌芽狀態。未來需要新的方法來發現更多有效的藥物,改善體內抗朊病毒作用的一種可能方法是聯合治療,例如:與自噬誘導劑和抑制從頭朊病毒轉化的化合物聯合處理可協同抗朊病毒作用。某些單一藥物常存在缺陷,如甲磺酸伊馬替尼穿透血腦屏障的效果較差,因此,另一種策略可以是在現有的藥物基礎上對藥物進行功能化修飾,促進藥物通過血腦屏障,進入到大腦中發揮作用。闡明自噬在朊病毒感染中的作用機制,將可行性方案應用到臨床治療,挽救患者的生病、延長患者的生存時間,是每一位醫療科研人員的共同目標。
6 總結
朊病毒病是一種由毒性蛋白(PrPSc)異常大量聚集所導致的嚴重神經退行性疾病,而PrPSc毒性蛋白多由中樞系統中正常的朊蛋白PrPC異常折疊形成。目前全球范圍內治療手段有限,患者2 年生存率較低。朊病毒相關的自噬機制的研究對于探尋延緩疾病進程及治療朊病毒病的新思路與有效方案具有重要意義。本文闡述了自噬在朊病毒病中通過TFEB 調控的ALP 途徑、mTOR 介導的自噬抑制途徑、AMPK 介導的ALP 途徑3 個主要機制來增加自噬通量、調節PrPSc在體內的聚集。ALP的缺陷會導致蛋白質聚集、有毒蛋白種類的產生和積累以及細胞器的功能失調,維持ALP 途徑的穩定可為治療帶來新希望。TFEB 在ALP 的3 種自噬途徑中起著至關重要的作用,如何調控TFEB 的表達或許可作為一個新的突破口。此外,靶向抑制mTOR 介導的自噬抑制途徑是誘導自噬、降低PrPSc聚合物的有效途徑。現有藥物中雷帕霉素是經典的mTOR 抑制劑,可有效延長小鼠存活時間,相較于甲磺酸伊馬替尼來說,雷帕霉素有可能順利通過血腦屏障,在中樞神經系統發揮作用,彌補甲磺酸伊馬替尼只在中樞神經系統外發揮作用的缺陷。目前在臨床階段,已經確定使用自噬誘導劑雷帕霉素治療可以通過靶向抑制mTOR 依賴的自噬抑制途徑來降低PrPSc的聚集水平。在自噬通量的檢測方面,LC3-Ⅱ和p62 蛋白的表達量是良好的觀測指標。LC3-Ⅱ下調、p62 蛋白上調均可說明自噬通量的減低,此時提示或許可將誘導自噬作為有效的治療途徑,為朊病毒病的治療帶來希望。同時,在誘導自噬時保持鈣的穩態和鈣介導的信號傳導、糾正不平衡的鈣蛋白酶-組織蛋白酶激活,可以更好地確保自噬功能的正常發揮,減輕鈣超載帶來的線粒體損傷等其他病理變化。以上3 種自噬在朊病毒病中的機制為臨床靶向抗朊病毒病藥物的研發提供了新思路。從目前研究結果來看,雷帕霉素與二甲雙胍是較為有效的藥物,其他基于以上機制的藥物還包括甲磺酸伊馬替尼、AR-12、白藜蘆醇、PPS、奎納克林、多西環素、剛果紅、馬來酸氟吡啶、硫磺素、鐵的四吡咯衍生物等。然而,過度誘導自噬可能會造成嚴重的后果,如何尋找自噬通量調控點、解決過度自噬帶來的不良反應,以及在嚴重感染后期藥物是否能持續發揮顯著效果、如何確定最佳的給藥量,都是未來科研工作者探索與努力的方向。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
中國衛生(2016年3期)2016-11-12 13:23:26
中國衛生(2014年12期)2014-11-12 13:12:52
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
七彩語文·畫刊(2012年3期)2012-04-29 00:00:00
