固相萃取-高效液相色譜法同時測定水體中甲萘威和阿特拉津
2022-02-19 03:36:54豆艷霞何云勇李會玲
浙江化工 2022年1期
關鍵詞:方法
豆艷霞,何云勇,李會玲,馮 莎
(1.安康市環境保護監測站,陜西 安康 725000;2.安康市漢濱區環境監測站,陜西 安康 725000)
甲萘威,又名西維因,化學名稱為1-萘基-N-甲基氨基甲酸酯,屬萘基氨基甲酸脂類農藥,廣泛應用于控制農作物、樹木和觀賞植物害蟲。阿特拉津,又名莠去津,化學名稱為2-氯-4-乙胺基-6-異丙胺基-1,3,5-三嗪,是一種廣泛使用的除草劑。這兩種農藥的廣泛應用,對水生態環境和人類飲用水造成潛在威脅[1-3]。《生活飲用水衛生標準》(GB 5749—2006)中規定阿特拉津的限值為0.002 mg/L,《地表水環境質量標準》(GB 3838—2002)中規定甲萘威和阿特拉津的限值分別為0.05 mg/L 和0.003 mg/L。
目前,在環境監測領域,針對水中甲萘威和阿特拉津測定的標準方法有液相色譜法(HPLC)和氣相色譜法(GC)[4-6]。此外,隨著檢測技術的發展,液相色譜-質譜法(LC-MS)、氣相色譜-質譜法(GC-MS)測定水中甲萘威、阿特拉津常有報道[7-9]。以上幾種分析方法都是將兩種物質分開檢測。本研究采用固相萃取-高效液相色譜法同時測定水中的甲萘威和阿特拉津。方法簡單快速、準確率高、精密度好、檢出限低,可用于地表水中甲萘威和阿特拉津的檢測。
1 材料與方法
1.1 儀器和試劑
儀器:賽默飛EVOLUTION 220 紫外-可見分光光度計;賽默飛U3000 液相色譜儀,SUPELCO 固相萃取儀;C18固相萃取柱;Organomation 氮吹儀。
試劑:甲萘威標準樣品(1000 mg/L,);阿特拉津標準樣品(100 mg/L);甲萘威質控樣((57.4±3.50)mg/L);阿特拉津質控樣((44.5±4.0)mg/L);二氯甲烷:色譜純;甲醇:色譜純;無水硫酸鈉:優級純,400 ℃灼燒4 h。
1.2 樣品前處理
1.3 色譜條件
C18色譜柱(250 mm×4.6 mm,5 μm),V(甲醇):V(水)=6:4,流速為0.8 mL/min,柱溫為40 ℃,紫外檢測器波長為222 nm。
2 結果與討論
2.1 前處理方法的確定
按照1.2 的方法,對添加標準溶液后的100 mL水樣進行固相萃取。同時按照HJ 587—2010 和GB/T 5750.9—2006 中前處理要求,分別對阿特拉津和甲萘威進行單獨萃取,結果見表1。從表1 可知,本試驗方法同時萃取甲萘威和阿特拉津,具有較高的萃取率和較好的精密度,萃取率分別為89.7%±5.1%和91.7%±3.3%,同時具有溶劑使用量少、操作人員暴露風險低等優點。

表1 幾種前處理方式的對比Tab.1 Comparison of different sample preparation methods
2.2 色譜條件的確定
2.2.1 紫外吸收波長的確定
(1)波長篩選
使用紫外/可見分光光度計在190~320 nm 波長范圍內分別掃描阿特拉津和甲萘威標準溶液,掃描結果見圖1、圖2。由圖可知,阿特拉津在222 nm 左右有明顯吸收峰,甲萘威分別在220 nm和280 nm 左右有明顯吸收峰。
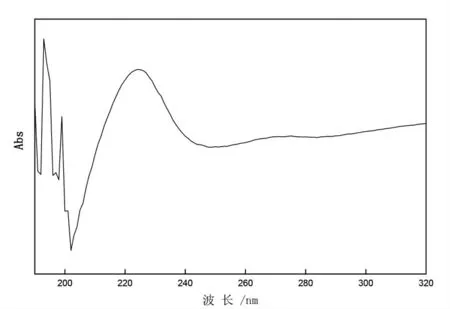
圖1 阿特拉津光譜掃描圖Fig.1 Ultraviolet absorption spectra of atrazine
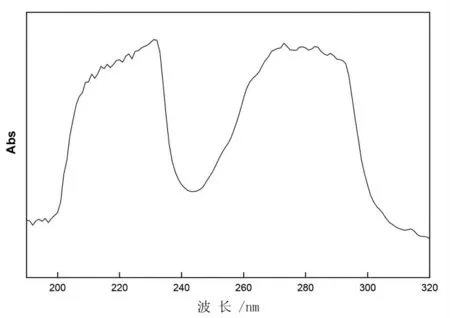
圖2 甲萘威光譜掃描圖Fig.2 Ultraviolet absorption spectra of carbaryl
(2)波長確定
根據以上掃描結果,向液相色譜儀中分別注入10 μL 的1 μg/L 阿特拉津、甲萘威標準溶液,檢測器波長在210~230 nm 范圍變化,得到不同色譜圖。以波長為橫坐標,兩種物質的響應值(mAu)為縱坐標作圖,見圖3。由圖3 可知,阿特拉津在222 nm 處有最大吸收波長,甲萘威在220 nm處有最大吸收波長,考慮同等條件下,阿特拉津吸收響應值較甲萘威弱,且甲萘威在220 nm 和222 nm 處吸收響應值相差不到5%,選取220 nm和222 nm 為兩種物質最佳吸收波長的研究均有報道[10-12]。故確定222 nm 為試驗波長。
對于中毒性僵苗田塊,要及時排水曬田,增溫補氧,改善土壤環境。堅持淺水勤灌與輕擱田相結合,提高土壤通透性,加速土壤環境更新,氧化還原性有毒物質。對于冷害僵苗的田塊,在秧苗返青后,也應排水露田,以水調溫,以水保溫,日曬夜灌,提高水溫和土溫。
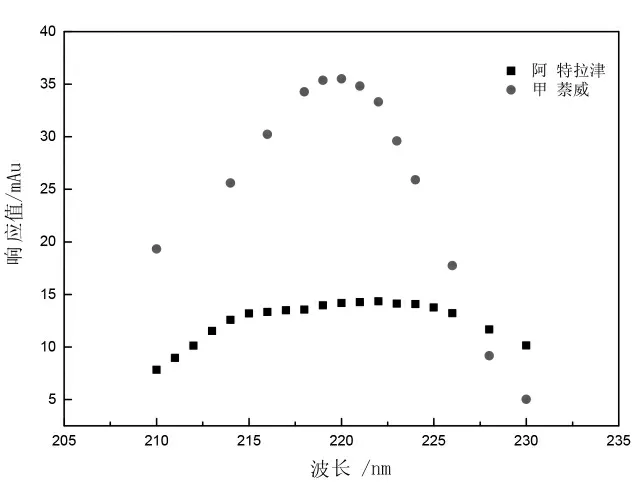
圖3 兩種物質在液相色譜不同檢測波長下的響應值Fig.3 Response values of two target compounds to different ultraviolet wavelength
2.2.2 流動相比例的確定
選取V(甲醇):V(水)為6:4、7:3 和8:2 進行流動相比例測試,不同流動相比例下兩種物質的色譜峰結果見表2,色譜圖見圖4。
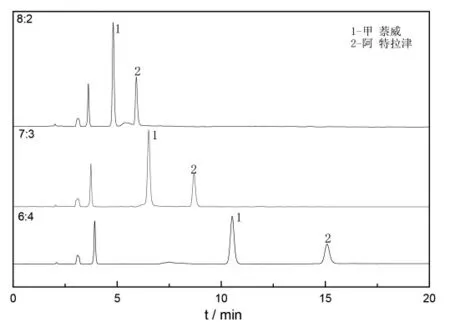
圖4 不同流動相比例下兩種物質的色譜圖Fig.4 Chromatogram of two target compounds with different proportion of mobile phase
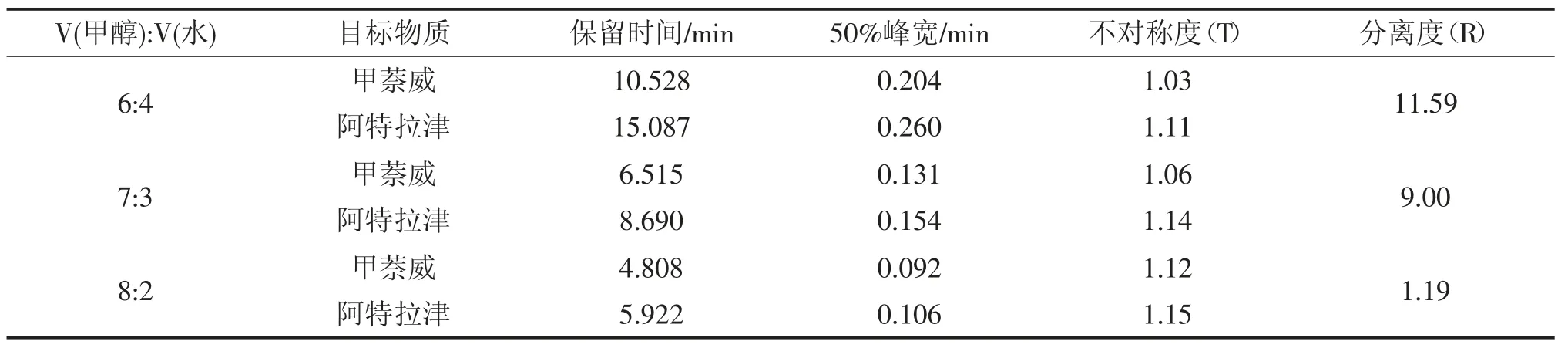
表2 不同流動相比例下兩種物質的色譜峰結果Tab.2 Peak parameters for two target compounds with different proportion of mobile phase
由表2 可知,隨著流動相比例的增加,兩種物質的保留時間均提前,可見在反相液相色譜中,增大流動相中甲醇的量,可以加快目標物質的出峰時間。隨著流動相比例的增加,兩種物質的峰寬減小、不對稱度增大,V(甲醇):V(水)為6:4時,甲萘威和阿特拉津色譜峰的不對稱度均最接近1(《中國藥典》規定T 值為0.95~1.05)。兩種物質的分離度隨著流動相比例的增加而減小,V (甲醇):V(水)為8:2 時,分離度(R)為1.19,分離效果較差(《中國藥典》規定值R 應大于1.5)。
由圖4 可知,V(甲醇):V(水)為8:2 時,兩種物質的主峰接近,中間有不完全分離部分;V(甲醇):V(水)為7:3 時,甲萘威主峰前后有不規則小峰出現;V(甲醇):V(水)為6:4 時,甲萘威和阿特拉津峰型均較好。本試驗確定流動相比例為6:4。
2.2.3 流動相流速的確定
選取1.2 mL/min、1.0 mL/min、0.8 mL/min 和0.6 mL/min 流動相流速進行測試,結果見表3。由表3 可知,流速主要影響物質的出峰時間,對兩種物質的分離度影響不大。本試驗選取流動相流速為0.8 mL/min,在實際工作中可根據需要適當提高流速,加快出峰時間。
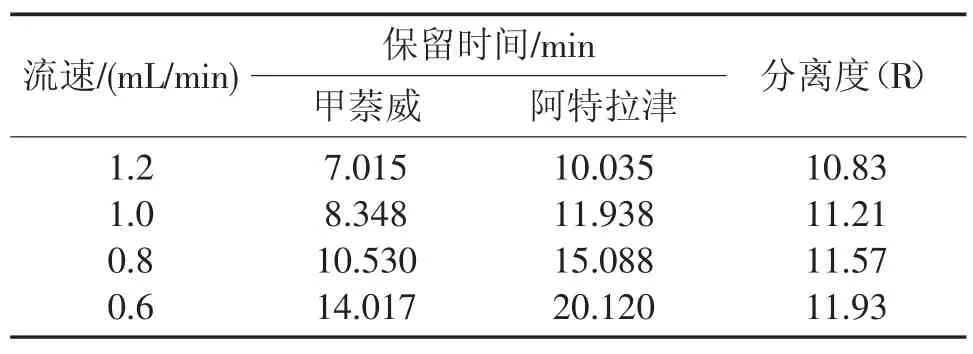
表3 不同流速下兩種物質的保留時間和分離度Tab.3 Retention times and resolution of two target compounds with different proportion of mobile phase
2.2.4 柱溫的選擇
選取20 ℃、30 ℃和40 ℃進行柱溫測試。柱溫升高,兩種物質出峰時間稍有提前(提前1.2~1.8 min),本試驗選取柱溫為40 ℃。
2.3 方法測試
2.3.1 線性范圍測試
用甲萘威和阿特拉津標液配置兩種物質的混合標準使用液,得到濃度分別為0.05 mg/L、0.1 mg/L、0.5 mg/L、1 mg/L、2 mg/L、5 mg/L 的標準系列。以濃度為橫坐標,峰面積為縱坐標,進行回歸計算,標準曲線結果見表4。結果表明,在0.05~5 mg/L濃度范圍內,兩種物質線性均良好。
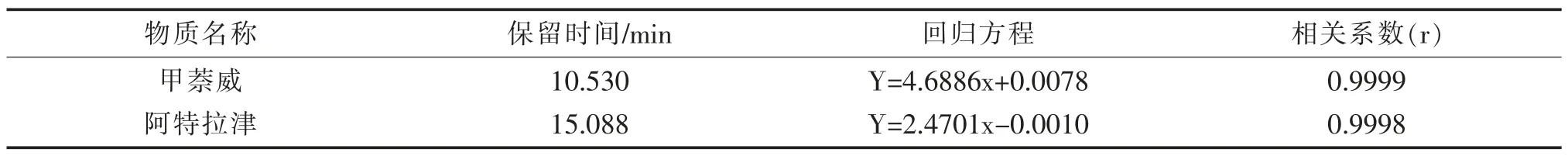
表4 兩種物質的回歸方程和相關系數Tab.4 Regression equations and correlation coefficients of two target compounds
2.3.2 檢出限測試
通過空白加標的方式確定方法檢出限,將低濃度的標準樣品加到純水中,得到7 個空白加標樣品,分析7 次,按照下式(1)計算檢出限。

式(1)中:S 為7 次測定的標準偏差;t(6,0.99)=3.143(查表得)。
本試驗方法確定的甲萘威、阿特拉津檢出限見表5,甲萘威和阿特拉津檢出限分別為0.00002 mg/L和0.00005 mg/L,均低于現有標準方法。

表5 兩種物質的檢出限Tab.5 The detection limit of two target compounds
2.3.3 準確度、精密度測試
分析有證標準物質測試方法的準確度和精密度,結果見表6。由表6 可知,該方法測定甲萘威和阿特拉津有證標準物質,準確度分別為97.4%和97.5%,精密度分別為1.1%和1.7%。
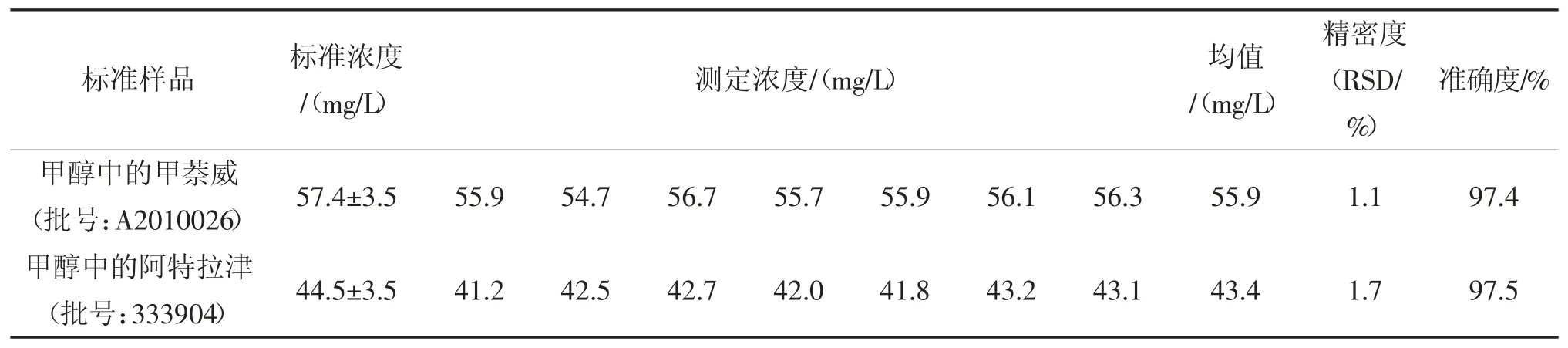
表6 本方法分析標準樣品的準確度和精密度Tab.6 Recoveries and RSDs of two target compounds in the certified reference materials
分析實際水樣測試方法的準確度和精密度,結果見表7。水樣采取加標方式制備,其中水樣1(低濃度)中甲萘威、阿特拉津加標量為0.1 μg;水樣2(高濃度)中甲萘威、阿特拉津加標量為1 μg。按照1.2 前處理步驟,萃取濃縮后上機測定。由表7 可知,甲萘威的回收率為75.4%~89.7%,相對標準偏差為5.10%~8.85%;阿特拉津的回收率為76.1%~91.7%,相對標準偏差3.26%~7.44%。
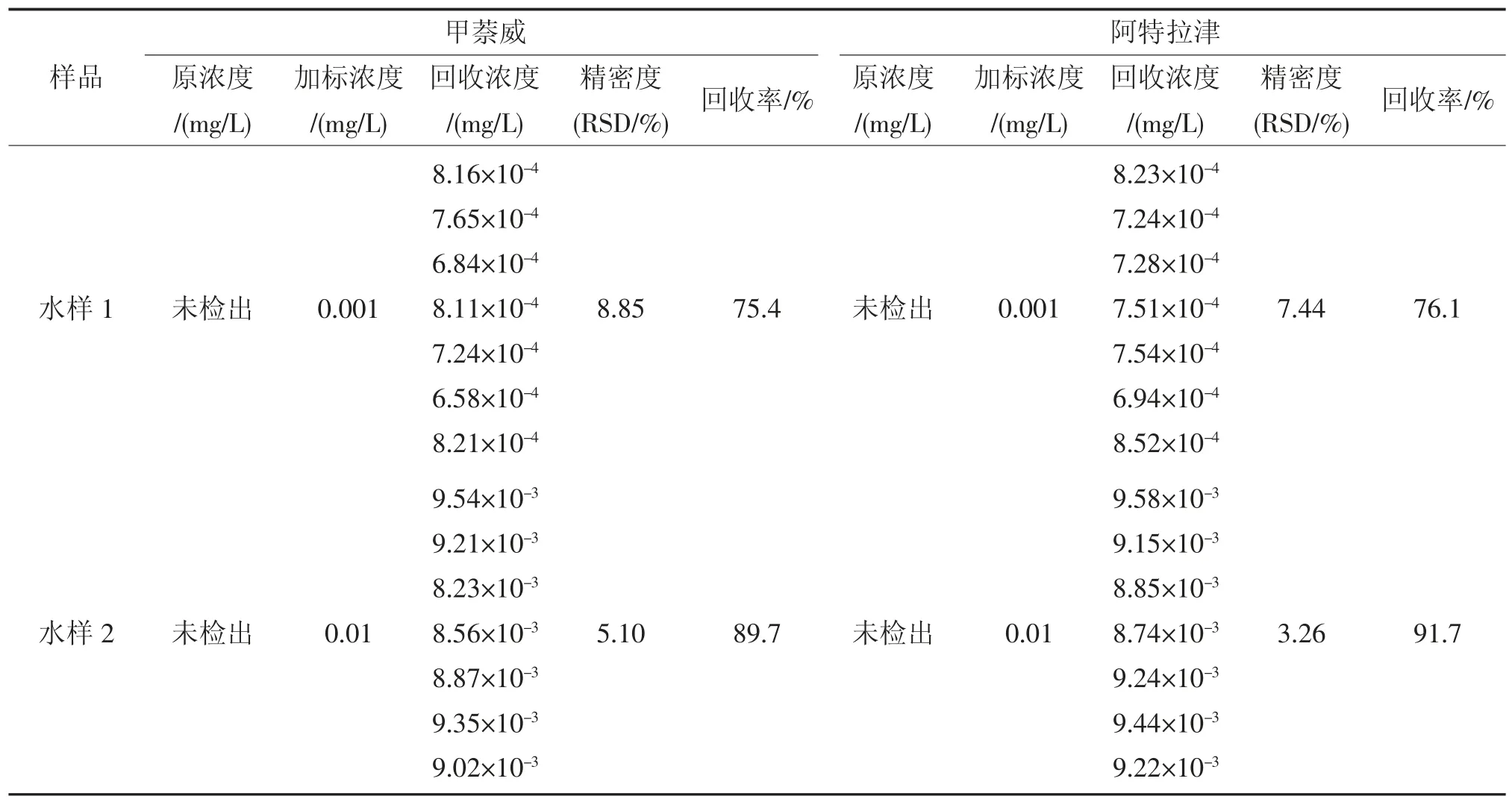
表7 本方法分析實際水樣的準確度和精密度Tab.7 Recoveries and RSD of two target compounds spiked in a real sample
3 結論
本研究采用固相萃取-高效液相色譜法同時測定水體中的甲萘威和阿特拉津。結果表明,使用固相萃取代替現有標準方法中的液液萃取具有較高的回收率,同時減少了萃取溶劑的使用量和操作人員暴露風險。通過色譜條件優化,該方法準確度高,線性和精密度良好,檢出限滿足需求,可用于同時測定水體中的甲萘威和阿特拉津。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
