酸溶ICP-AES法測定鋁硅系耐火材料中8種次量元素
2022-01-08 07:53:10孟麗麗
山東冶金 2021年6期
關(guān)鍵詞:實(shí)驗(yàn)分析
孟麗麗
(山東省冶金科學(xué)研究院有限公司,山東 濟(jì)南 250014)
1 前言
目前耐火材料中8種次量元素含量的化學(xué)分析均采用國家標(biāo)準(zhǔn)方法,其中CaO采用火焰原子吸收光譜法和EDTA容量法;MgO采用二甲苯胺藍(lán)Ⅰ-溴化十六烷基三甲銨光度法和火焰原子吸收光譜法;K2O和Na2O采用火焰原子吸收光譜法;P2O5采用鉍磷鉬藍(lán)光度法測定;Fe2O3采用鄰二氮雜菲分光光度法;而對于Li2O 和Sr2O 元素,目前尚無標(biāo)準(zhǔn)測定方法。以上各方法分析時間長,步驟繁瑣。本文研究了用酸溶溶解樣品ICP-AES 法測定鋁硅系耐火材料中CaO、MgO、K2O、Na2O、P2O5、Fe2O3、Li2O、Sr2O 元素含量的新方法,確定了最佳的實(shí)驗(yàn)及測定條件,顯著提高了分析的準(zhǔn)確度、精密度和分析速度[1-3]。
2 實(shí)驗(yàn)部分
2.1 儀器及工作參數(shù)
電感耦合等離子體發(fā)射光譜儀。儀器工作參數(shù):等離子體功率1 400 W;氬氣壓力0.8 MPa;霧化器流量0.80 L/min;冷卻氣流量13.00 L/min;輔助氣流量1.00 L/min;積分時間30 s。電子天平(BSA224S-CW);純水機(jī)(H2O-MM-UV-T)。
2.2 試 劑
鹽酸(ρ1.19 g/mL)、硝酸(ρ1.42 g/mL)、高氯酸(ρ1.67 g/mL)、氫氟酸(ρ1.15 g/mL);高純氬(體積分?jǐn)?shù)>99.99%);實(shí)驗(yàn)室用酸為優(yōu)級純(GR),實(shí)驗(yàn)用水為高純?nèi)ルx子水。
2.3 標(biāo)準(zhǔn)儲備溶液
鈣標(biāo)準(zhǔn)溶液1 00 μg/mL[GBW(E)081277];鎂標(biāo)準(zhǔn)溶液1 00 μg/mL[GBW(E)081287];鉀標(biāo)準(zhǔn)溶液100 μg/mL[GBW(E)081285];鈉標(biāo)準(zhǔn)溶液100 μg/mL[GBW(E)081290];磷標(biāo)準(zhǔn)溶液100 μg/mL[GBW(E)081293];鋰標(biāo)準(zhǔn)溶液100 μg/mL[GBW(E)081286];鍶標(biāo)準(zhǔn)溶液100 μg/mL[GBW(E)081301]。
2.4 實(shí)驗(yàn)方法
2.4.1 實(shí)驗(yàn)條件選擇
(1)儀器測量條件。選擇混合標(biāo)準(zhǔn)溶液,進(jìn)行了等離子體功率、霧化器流量、積分時間、泵速、霧化器壓力等實(shí)驗(yàn)。實(shí)驗(yàn)表明,隨著霧化壓力的減小,各分析元素強(qiáng)度增加變化不明顯,霧化氣壓力過低,穩(wěn)定性變差;霧化器流量增大元素強(qiáng)度明顯降低,故選擇0.80 L/min最佳。泵速減小強(qiáng)度降低,泵速增加強(qiáng)度升高,在2.00 mL/min為最好。積分時間在5~40 s,各分析元素的強(qiáng)度變化不明顯,30 s時最好。RF 功率分別在1 200、1 300、1 400、1 500、1 600 W進(jìn)行實(shí)驗(yàn)。結(jié)果顯示:低功率時,各元素強(qiáng)度均降低很多;功率過高時強(qiáng)度升高但穩(wěn)定性差。綜合考慮穩(wěn)定性、靈敏度和測定元素的相關(guān)性,確定儀器測量條件為:等離子體功率1 400 W;氬氣壓力0.8 MPa;霧化器流量0.80 L/min;冷卻氣流量13.00 L/min;輔助氣流量1.00 L/min;積分時間30 s。
(2)溶樣酸及酸度。試樣采用鹽硝酸、氫氟酸分解后,冒高氯酸煙至近干后加鹽酸溶解。對溶樣酸鹽酸、氫氟酸、硝酸、高氯酸的用量進(jìn)行了實(shí)驗(yàn)。本方法實(shí)驗(yàn)加入3、5、10、15、20 mL,實(shí)驗(yàn)發(fā)現(xiàn),硝酸和高氯酸用量多少IR 值無明顯波動,鹽酸和氫氟酸加入3 mL 和5 mL 時樣品溶解不完全,10 mL、15 mL 溶解完全時IR 值無明顯區(qū)別,20 mL 時引入的空白大。考慮到酸用量10 mL 以下樣品溶解不完全及用量20 mL 時,會引入所測元素的試劑空白,而且由于酸度過大譜線強(qiáng)度減弱,故采用10~15 mL 即可。本實(shí)驗(yàn)用酸量為13 mL 鹽酸、硝酸、氫氟酸,且分兩次加入溶解樣品。
(3)樣品處理。將試料置于鉑皿中(若無鉑皿,也可用聚四氟乙烯燒杯),用少量水潤濕,沿壁加入10 mL 鹽酸,調(diào)壓150 V 于電熱板上加熱溶解30 min,取下稍冷加入5 mL 硝酸、10 mL 氫氟酸、5 mL高氯酸,加熱分解至冒盡白煙。取下,稍冷,用水沖洗鉑皿(聚四氟乙烯燒杯)壁。加入3 mL 鹽酸、3 mL 硝酸、3 mL 氫氟酸、3 mL 高氯酸、3 mL 氫氟酸,繼續(xù)加熱至白煙冒盡,取下,冷卻。加入3 mL 鹽酸,沿壁吹水加熱溶解鹽類。取下冷卻至室溫后移入50 mL 容量瓶中,以水稀釋至刻度混勻靜置(干過濾)備用,隨同試樣做空白實(shí)驗(yàn),在ICP-AES(電感耦合等離子體發(fā)射光譜儀)上進(jìn)行測定。
2.4.2 工作曲線繪制
于空白試液中加入標(biāo)準(zhǔn)溶液,在選定的最佳儀器條件下將系列標(biāo)準(zhǔn)溶液引入等離子體中,運(yùn)行分析程序,建立工作曲線,以譜線強(qiáng)度值為y軸,分析元素的濃度為x軸,由儀器配套的計(jì)算機(jī)程序軟件自動給出線性相關(guān)系數(shù)(>0.999),系列溶液工作曲線見表1,工作曲線繪制見表2。

表1 系列工作曲線 μg/mL
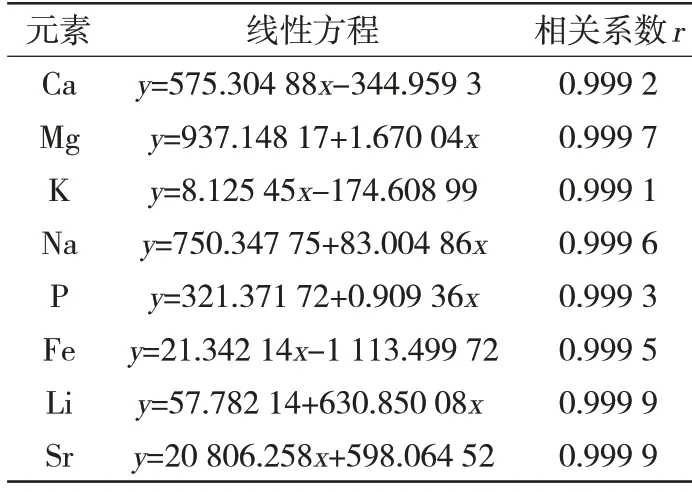
表2 工作曲線繪制
3 結(jié)果與討論
3.1 基體效應(yīng)的消除
試液基體的不同將導(dǎo)致結(jié)果不一致,基體對譜線強(qiáng)度有影響,一般會疊加加強(qiáng)。故配制混合標(biāo)準(zhǔn)溶液時,應(yīng)稱取與試樣中含量接近高純鋁進(jìn)行基體匹配,有效地消除基體效應(yīng)。
3.2 分析譜線的選擇
在發(fā)射光譜儀的譜線庫中查看各元素的強(qiáng)度信息和譜線干擾,根據(jù)樣品的含量高低,選擇強(qiáng)度適宜的譜線。將所測定的系列工作曲線標(biāo)液進(jìn)行譜圖疊加,選擇干擾少的譜線作為分析譜線,并進(jìn)行了背景校正,故本法選擇的最佳分析譜線波長。分析時,為使分析結(jié)果的可靠性更好一些,可選一條分析譜線,也可選兩條譜線進(jìn)行分析對比。具體分析線波長的選擇結(jié)果見表3。
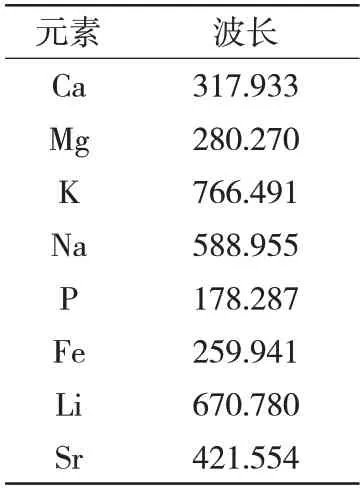
表3 分析線波長 nm
3.3 檢出限及測定下限
空白進(jìn)行了10 次測量,得到檢測結(jié)果見表4。以試劑空白溶液10 次測定結(jié)果的3 倍標(biāo)準(zhǔn)偏差作為檢出限,10倍標(biāo)準(zhǔn)偏差作為方法的測定下限。

表4 檢出限及測定下限 %
4 精密度、準(zhǔn)確度及回收率實(shí)驗(yàn)
4.1 正確度和精密度
對美國NIST 編號為77a 的耐火材料標(biāo)準(zhǔn)物質(zhì)進(jìn)行10 次測量,結(jié)果如表5 所示,計(jì)算各元素的相對標(biāo)準(zhǔn)偏差(RSD),證明本實(shí)驗(yàn)室運(yùn)用該標(biāo)準(zhǔn)檢測具有較高的正確度和精密度。
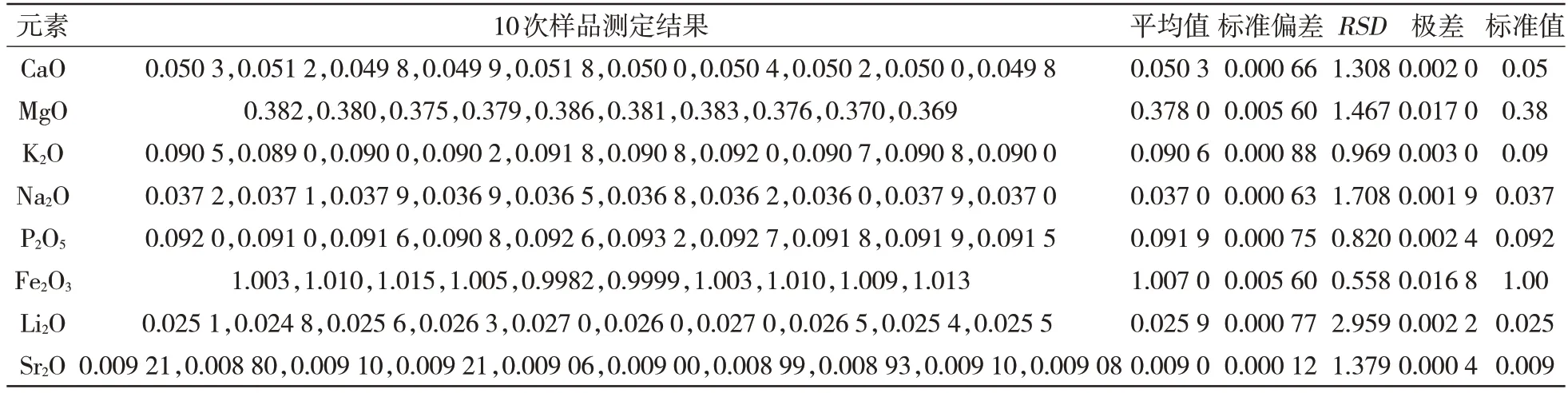
表5 正確度和精密度實(shí)驗(yàn) %
4.2 回收率
以77a 的耐火材料標(biāo)準(zhǔn)物質(zhì)為基體,分別加入標(biāo)準(zhǔn)溶液,按照標(biāo)準(zhǔn)進(jìn)行測定,分析結(jié)果如表6所示。
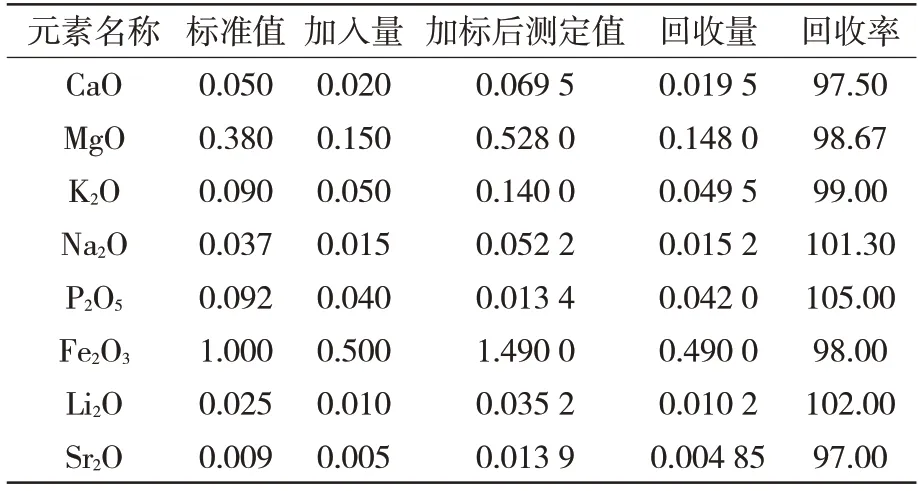
表6 回收率實(shí)驗(yàn) %
通過加標(biāo)回收實(shí)驗(yàn)計(jì)算各元素的回收率在97%~102%,符合95%~105%的基本要求,證明本實(shí)驗(yàn)室運(yùn)用該標(biāo)準(zhǔn)檢測回收率良好。
5 結(jié)語
(1)在選定的條件下,對熔劑用量、浸出酸選擇及用量、譜線的選擇、基體的干擾做了大量實(shí)驗(yàn),對鋁硅系耐火材料中的8 種次量元素進(jìn)行了同時測定。
(2)本方法具有靈敏度高、試劑用量少、線性范圍寬等優(yōu)點(diǎn)。方法檢出限低,精密度高,結(jié)果準(zhǔn)確可靠。縮短了檢測時間,提高了工作效率。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
民用飛機(jī)設(shè)計(jì)與研究(2020年4期)2021-01-21 09:15:02
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
電子制作(2018年18期)2018-11-14 01:48:24
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:22
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
中國中醫(yī)藥現(xiàn)代遠(yuǎn)程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
