荷花腐敗病菌的熒光定量PCR檢測
2021-12-13 08:52:22溫華強舒燦偉曾莉莎周而勛
中國農學通報 2021年34期
關鍵詞:檢測
溫華強,舒燦偉,曾莉莎,周而勛
(1華南農業大學植物保護學院/廣東省微生物信號與作物病害防控重點實驗室,廣州 510642;2東莞市農業科學研究中心,廣東 東莞 523086)
0 引言
荷花是一種重要的經濟作物,其地下莖(藕)及種子(蓮子)均可食用,其花具重要的觀賞價值。中國荷花栽培歷史悠久,除西藏和青海外,各地均有種植,是世界荷花的栽培中心[1]。近年來,在廣東珠三角的荷花栽培集中區爆發了大范圍的荷花腐敗病,嚴重制約了荷花產業及旅游業的發展[2]。荷花腐敗病又稱腐爛病、枯萎病,是荷花種植區發生最嚴重、最為普遍的病害之一[3]。該病害具有爆發速度快、破壞性強等特征,在整個荷花生長周期內均可發生,如果無有效的防治措施和防治藥物,能夠造成20%的減產,嚴重的可達60%以上,甚至絕收[4-5]。2014年,Zhu等[6]鑒定了荸薺枯萎病,認為國內的荸薺枯萎病病原菌為普通鐮刀菌(Fusarium commune)及一個未定名的鐮孢菌(Fusariumsp.)。2016年,Zhu等[7]建立了檢測土壤及植物組織中荸薺枯萎病菌(F.commune)含量的熒光定量PCR檢測方法。2017年,曾莉莎等[8]對廣東、廣西、江西、福建、湖南、湖北等地的荷花腐敗病病原菌進行鑒定及系統發育研究,通過形態學和分子鑒定,認為普通鐮刀菌(F.commune)是引起荷花腐敗病的病原菌,并且與分離自國內荸薺枯萎病的病原菌為同種。
引起荷花腐敗病的鐮刀菌是一種土壤習居菌,其菌絲體和厚垣孢子可以在土壤長期存活,成為來年的再侵染源[9]。荷花為多年生水生植物,荷花腐敗病的發病程度與水底土壤中的荷花腐敗病菌含量密切相關。對此,掌握荷花腐敗病發生程度和土壤中荷花腐敗病菌的數量的關系,將有助于在荷花種植前后有針對性地防治該病害。然而,傳統的檢測鐮刀菌的方法,比如平板稀釋法,對其進行分離、形態學以及致病性鑒定等,步驟繁瑣、耗時較長,當病害爆發時,由于診斷緩慢,往往延誤最佳防治時機,很容易造成嚴重的損失[10-12];常規PCR相對來說速度較快、特異性高,但檢測靈敏度仍有待提高[13];對土壤中的病原菌進行實時定量檢測可為制定科學的病情防控方案提供更加科學的依據[14]。本研究以此為切入點,聯合東莞市香蕉蔬菜研究所技術人員對該病害的發展動態進行監測,旨在研發一種快速、準確的定量檢測荷花腐敗病菌的熒光定量PCR技術,對東莞蓮湖的土壤、蓮藕等樣品的荷花腐敗病菌數量進行動態監測,旨在為該病害的有效防治提供重要依據。
1 材料與方法
1.1 荷花腐敗病樣品的采集與病原菌分離
從廣東省東莞市橋頭鎮蓮湖的荷花腐敗病發病區采集病藕組織,通過常規分離、培養、純化和鑒定,獲得荷花腐敗病菌(F.commune)純凈菌株,用于本研究的所有實驗。
1.2 荷花腐敗病區土壤中的荷花腐敗病菌基因組DNA的提取
土壤的基因組DNA使用E.Z.N.A.?Soil DNA kit(Omega Bio-Tek,USA)試劑盒提取。
1.3 荷花腐敗病菌菌絲基因組DNA的提取
將荷花腐敗病菌菌株接種于200 mL的PDB培養基,置于25℃、180 r/min條件下的搖床上培養5~8天。將搖床培養的PDB培養基取出,用抽濾機抽濾,得到荷花腐敗病菌的菌塊(含菌絲和孢子),用滅菌的濾紙吸干水分,置于-80℃下保存。荷花腐敗病菌DNA的提取使用E.Z.N.A.?High Performance(HP)Fungal DNA Kit(Omega Bio-Tek,USA)試劑盒。
1.4 各種發病級別荷花病組織基因組DNA的提取
各種發病級別荷花組織基因組DNA的提取使用E.Z.N.A.?High Performance(HP)Plant DNA(Omega Bio-Tek,USA)試劑盒。
1.5 qPCR引物的選用
由于本研究的荷花腐敗病菌與國內報道的荸薺枯萎病菌均為普通鐮刀菌(F.commune)[8,12],本研究采用的引物是朱志賢[15]篩選的、對荸薺枯萎病菌具有高度特異性的一對引物FO1(5’-GCCATAGGTCAGATAAC CAGTT-3’)和 FO2(5’-TCACTACTGGTGTCAGAAA CGG-3’),這對引物由北京擎科新業生物技術有限公司合成。
1.6 qPCR標準曲線的制作及靈敏度檢測
1.6.1 qPCR擴增的體系及使用材料和儀器 實驗的qPCR采用SYBR Green I熒光染料,PCR擴增體系為20μL,每份體系包含10.0μL ChamQ Universal SYBR qPCR Master Mix,10 μmol/L(引物工作濃度)的引物各0.4μL(FO1和FO2),1~2μL的DNA模板,加入RNase-free ddH2O至20μL。qPCR擴增的條件如下:95℃預變性 30 s;95℃變性 10 s,60℃延伸 30 s;95℃15 s,60℃ 60 s,95℃ 15 s。進行qPCR反應所用的儀器是Bio-Rad CFX96TM(Bio-Rad Laboratories,CA),獲得的數據使用Bio-Rad CFX Manager 3.1(admin)軟件進行分析
1.6.2 荷花腐敗病菌基因組DNA標準曲線的制作及靈敏度檢驗 將荷花腐敗病菌的基因組DNA稀釋至10 ng/μL,再以10倍濃度梯度稀釋荷花腐敗病菌的基因組DNA,即稀釋后的濃度依次為10 ng/μL、1 ng/μL、100 pg/μL、10 pg/μL、1 pg/μL、100 fg/μL、10 fg/μL、1 fg/μL。運行qPCR程序后收集數據,根據Ct值的含義,即每個反應管內的熒光信號到達設定的域值時所經歷的循環數,認為具有Ct值的為陽性樣本,沒有Ct值的是為陰性樣本,靈敏度為與陰性樣本相鄰的最后一個陽性樣本的濃度值。
1.6.3 加入植物組織DNA后荷花腐敗病菌基因組DNA標準曲線的繪制及靈敏度檢測 為研究在自然狀態下,荷花腐敗病菌侵染健康的荷花組織時,荷花的基因組DNA是否會對荷花腐敗病菌DNA的擴增造成影響,實驗中提取健康的荷花基因組DNA,稀釋至10 ng/μL,再吸取1μL加入到上述10倍濃度梯度的荷花腐敗病菌的基因組DNA配的20μL qPCR體系中(1μL荷花腐敗病菌的基因組DNA模板和1μL健康的荷花組織基因組DNA)進行擴增,繪制其標準曲線運行qPCR程序后收集數據,根據Ct值的含義,即每個反應管內的熒光信號到達設定的域值時所經歷的循環數,認為具有Ct值的為陽性樣本,沒有Ct值的是為陰性樣本,靈敏度為與陰性樣本相鄰的最后一個陽性樣本的濃度值。
1.6.4 接種土壤中荷花腐敗病菌標準曲線的繪制及靈敏度檢測 將荷花腐敗病菌菌株接種于200 mL的PDB培養基,置于25℃、180 r/min條件下的搖床上培養5~8天。準備滅過菌的紗布,用6層滅過菌的紗布過濾出荷花腐敗病菌的孢子,并將過濾好的孢子懸液轉移至15 mL離心管,10000×g離心15 min,用去離子滅菌水重復洗滌2~4次,至懸液顏色由黃色變為無色(或接近無色)。將分生孢子懸液置于顯微鏡下,利用血球板計數法,加入去離子蒸餾水稀釋成4×107、4×106、4×105、4×104、4×103、4×102、4×101、4個/mL等10倍濃度梯度的孢子懸液。將采自東莞橋頭鎮蓮湖的發病區土壤樣本去除雜物,置于干熱滅菌箱中,90℃、5 h滅菌烘干3次至恒重,利用研缽將其磨碎,稱取800 mg的滅菌土壤至15 mL離心管中,依次將一系列濃度梯度的分生孢子懸液取800μL加至上述的15 mL離心管中,以未接種孢子懸液的滅菌土壤作空白對照,使用E.Z.N.A.?Soil DNA kit(Omega Bio-Tek,USA)試劑盒對其提取基因組DNA,對其進行qPCR擴增和繪制標準曲線,認為具有Ct值的為陽性樣本,沒有Ct值的是為陰性樣本,靈敏度為與陰性樣本相鄰的最后一個陽性樣本的濃度值。
1.7 不同發病級別荷花植株中荷花腐敗病菌的qPCR檢測
在東莞橋頭蓮湖采集依次為無病、輕度發病、重度發病3個發病級別的荷花植株(病情分級標準由東莞橋頭工作人員制定)各3株,利用隨機取樣的方法在各種不同的發病級別的荷花植株中取3支莖稈,用5%的次氯酸鈉將其浸泡消毒5 min,接著用去離子滅菌水沖洗2~4次,用滅菌的吸水紙吸干表面水分。用滅菌處理過的剪刀依次從3支莖稈中部剪取一小段約100 mg的莖稈組織,提取其基因組DNA,進行qPCR擴增來檢測各種發病級別的荷花植株中荷花腐敗病菌的含量(根據測得的DNA含量計算),3次平行重復實驗。各種發病級別荷花植株的莖稈中的荷花腐敗病菌基因組DNA含量,是根據加入10 ng健康荷花組織DNA繪制的10倍濃度梯度的荷花腐敗病菌的基因組DNA的標準曲線獲得。
1.8 不同發病級別荷花植株根部土壤中荷花腐敗病菌的qPCR檢測
從各種發病級別的荷花植株的根部土壤處采集3份土壤樣品,每份大約1 kg。先將土壤樣品去除雜質,再將其置于干熱滅菌箱中,打開鼓風功能,38℃、24 h風干。提取處理后的土壤樣品的DNA,再進行qPCR擴增,得到相應的Ct值,根據接種荷花腐敗病菌的土壤中的荷花腐敗病菌基因組DNA的標準曲線,得到各種發病級別荷花植株的根部土壤中荷花腐敗病菌的孢子濃度。
1.9 數據分析
每一次的qPCR擴增反應由Bio-Rad CFX Manager 3.1(admin)軟件計算分析給出Ct值。10倍濃度梯度的荷花腐敗病菌的基因組DNA的標準曲線、加入植物組織DNA后荷花腐敗病菌基因組DNA的標準曲線和接種荷花腐敗病菌的土壤中的荷花腐敗病菌基因組DNA的標準曲線用Microsoft Excel(office 2016)軟件制作。用SPSS 22.0軟件的協方差分析檢驗,對是否添加健康荷花組織基因組DNA的一系列濃度梯度的荷花腐敗病菌的基因組DNA制作的2條標準曲線之間的截距,以及斜率是否存在顯著性差異。用相關分析方法分析各種發病級別的荷花植株中包含的荷花腐敗病菌的基因組DNA,以及各種發病級別荷花植株的根部土壤中包含的荷花腐敗病菌的基因組DNA與它們的發病級別之間的關聯。
2 結果與分析
2.1 qPCR的標準曲線及檢測靈敏度
用一系列濃度梯度的荷花腐敗病菌基因組DNA進行Real-Time PCR反應,得到的反應曲線如圖1所示。利用Microsoft Excel軟件制作的荷花腐敗病菌基因組DNA的標準曲線(圖2)為Y=-4.2734X+36.892(R2=0.9977),其中橫坐標X表示稀釋的一系列濃度梯度的DNA的濃度的對數,縱坐標Y表示與每個DNA濃度X值相對應的Ct值,R2=0.9977表明該標準曲線具有良好的線性關系。通過對qPCR擴增曲線(圖1)分析可得,qPCR的最低檢測限度為1 fg/μL。對該qPCR反應的溶解曲線(圖3)進行分析,擴增的產物僅有一個峰值(78.5℃),表明該qPCR擴增的DNA產物具有高度的特異性。
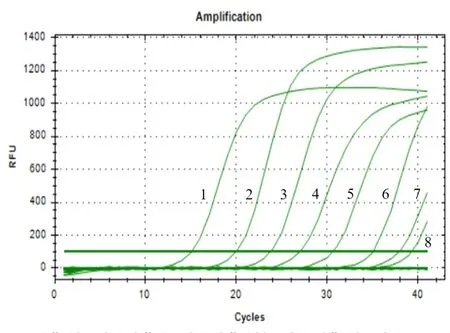
圖1 10倍濃度梯度的荷花腐敗病菌基因組DNA的qPCR擴增曲線
2.2 加入植物組織DNA后荷花腐敗病菌基因組DNA的標準曲線及檢測靈敏度
在添加健康荷花組織的基因組DNA后制作的一系列濃度梯度荷花腐敗病菌的DNA標準曲線(圖2)為Y=-3.6419X+34.501(R2=0.9939)。對添加了健康荷花組織的基因組DNA后的qPCR進行最低檢測限度分析,該qPCR反應的最低檢測限度為10 pg/μL。利用協方差分析檢驗2條標準曲線發現截距的差異性不顯著(P>0.05),但當比較斜率時則發現2條曲線的差異性顯著(P=0.01<0.05)。造成以上2條標準曲線之間存在差異的因素在于添加健康的荷花組織的基因組DNA,故采用添加了健康荷花組織的基因組DNA后制成的標準曲線計算發病的荷花組織中的荷花腐敗病菌的DNA含量。
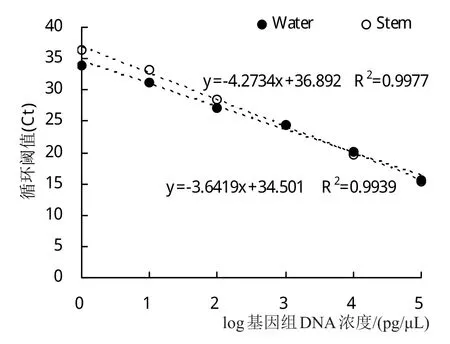
圖2 SYBR Green I PCR擴增基因組標準曲線
2.3 接種土壤中荷花腐敗病菌的標準曲線及靈敏度檢測
提取濃度依次為4×107、4×106、4×105、4×104、4×103、4×102、4×101、4、0 個分生孢子/(g·土)的基因組DNA進行qPCR反應,沒有接種孢子的滅菌土壤為對照組(圖4)。制成的標準曲線中坐標軸X表示分生孢子在土壤中的數目的對數,坐標軸Y表示其對應的Ct值,兩者的線性關系為Y=-3.1995X+34.262(R2=0.9911)(圖5)。由圖4可知,當分生孢子濃度低于4×102個分生孢子/(g·土)時不能被檢測出來,即最低檢測限度為4×102個分生孢子/(g·土)。
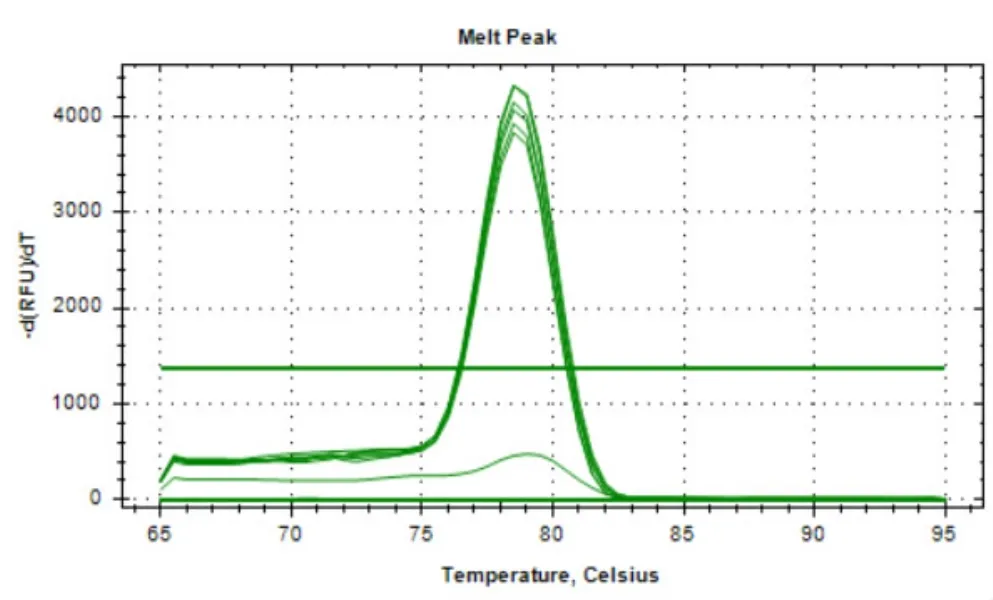
圖3 SYBR Green I PCR熔解曲線
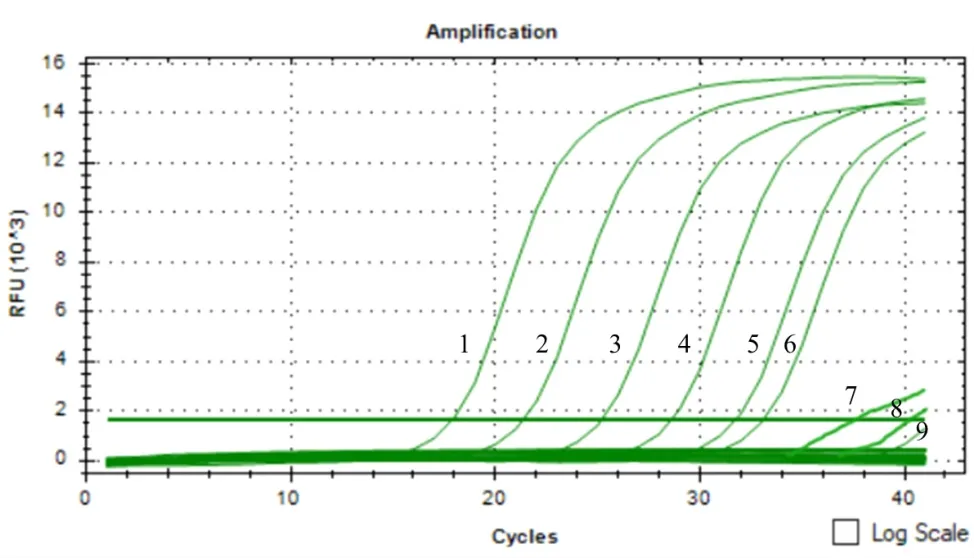
圖4 接種土壤的SYBR Green I PCR擴增曲線
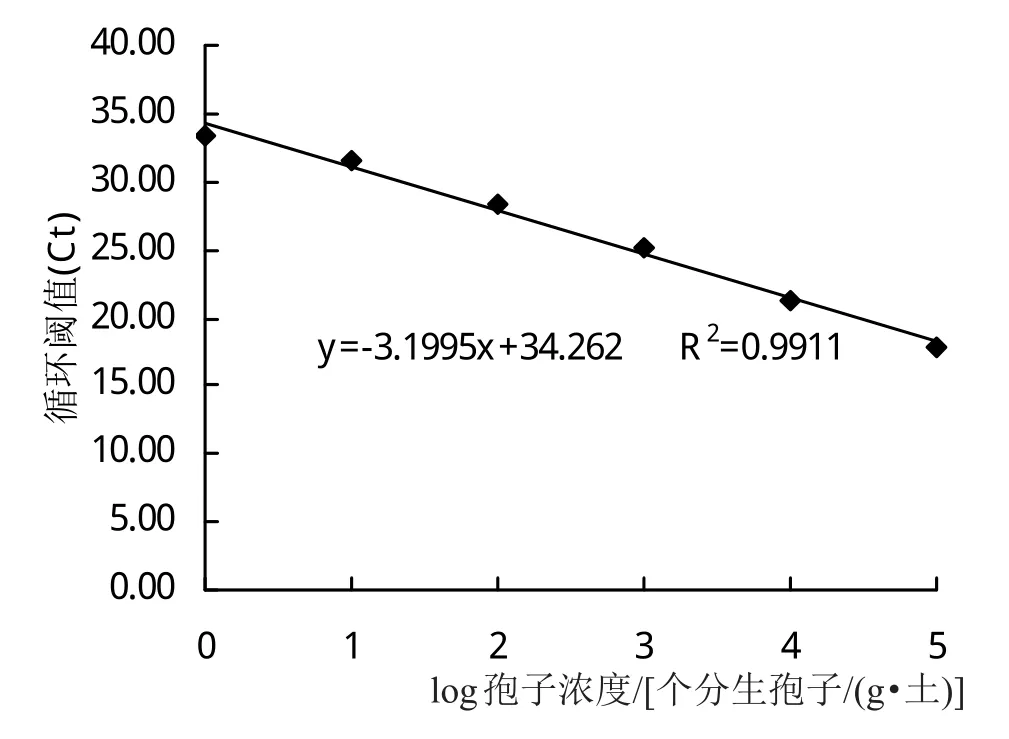
圖5 接種荷花腐敗病菌的土壤中的荷花腐敗病菌基因組DNA的標準曲線
2.4 各種發病級別荷花植株中荷花腐敗病菌含量與病害發生程度的相關性
用qPCR方法對各種發病級別的荷花植株中荷花腐敗病菌的含量進行了檢測。數據顯示,各種發病級別荷花植株樣品中荷花腐敗病菌DNA的含量依次為(0.010±0.003)、(0.0827±0.053)、(1.432±0.439)ng(表1)。其中,2級的DNA含量為0級的143.2倍。染病植株中荷花腐敗病菌DNA的含量與其發病程度呈正相關,皮爾森相關系數為0.927,顯著性(雙尾)為0.044。
2.5 各種發病級別植株地下土壤中荷花腐敗病菌含量與病害發生程度的相關性
0~2級發病級別植株的地下土壤中荷花腐敗病菌的含量依次為(3.618±2.284)×103、(34.400±12.891)×103、(164.733±54.270)×103個分生孢子/(g·土)。皮爾斯相關系數為0.819,顯著性(雙尾)為0.389,表明發病的荷花植株地下土壤荷花腐敗病菌的含量與病害發生程度呈正相關(相關度高),但顯著性較低。
3 結論與討論
隨著人們對荷花的觀賞需求和食用需求日益增加,荷花產業發展前景好。然而,素有“荷花癌癥”之稱的荷花腐敗病卻成了荷花產業發展的最大障礙之一,這種土傳性鐮刀菌病害侵染源是病殘體、病土等攜帶的病菌,包括菌絲體、厚垣孢子等鐮刀菌的越冬形態。據報道,輪作8年的土壤仍有菌絲和孢子能存活下來,并再度侵染健康的植株,所以仍然不能消除該病害的發生[16]。因此,研發一種能夠快速、有效地檢測土壤和罹病組織中荷花腐敗病菌含量的方法,對于荷花產業的健康發展具有重要的指導意義。
本研究利用SYBR Green I qPCR技術檢測了感染荷花腐敗病的荷花植株及其地下土壤中含有的荷花腐敗病菌的數量,其中qPCR方法的最低檢測限度是1 fg/μL,添加了健康的荷花植株的DNA后最低檢測限度是10 pg/μL,將分生孢子接種到經滅菌處理的土壤后的最低檢測限度是400個孢子/(g·土)。存在于植物或土壤中的PCR抑制因子,對qPCR檢測具有限制作用[17-18]。相比普通PCR,實時熒光定量PCR具有靈敏度高、精確定量、實時檢測擴增產物量的變化等諸多優點,更加適合用來動態監測荷花腐敗病菌的含量與發病程度的關系,分析荷花腐敗病菌的含量與發病級別的相關性。荷花腐敗病的初侵染源是帶菌的種藕和土壤,最初的發病部位是地下莖部,不易觀察,常失去最佳防治適期[5]。qPCR具有諸多優點,能夠在病害發生的初期進行快速檢測,而傳統的檢測手段難以做到,故已被廣泛地應用于病原物的快速檢測[19-21]。實時熒光定量PCR技術被廣泛應用于土壤中鐮刀菌引起的植物病害的快速診斷,陳利達等[22]根據鐮刀菌屬的TEF-1α基因構建的qPCR檢測體系,其靈敏度比常規PCR高104倍;王瑜等[23]根據腐皮鐮刀菌(F.solani)的tef 1基因,建立了腐皮鐮刀菌的SYBR Green qPCR檢測技術,其對重組質粒標準樣品的檢測靈敏度(1.0×102copies/μL)和穩定性(變異系數0.96%~1.68%)均優于常規PCR。李文學等[24]根據GenBank中葡萄霜霉病菌的cox2基因,建立并優化qPCR反應體系,該體系的檢測靈敏度是普通PCR檢測靈敏度的100倍,可達0.1 pg/μL。 Megan Ceris Matthews 等[25]利 用 SYBR green PCR技術對香蕉枯萎病菌(F.oxysporumf.sp.cubense,Foc)進行檢測,結果表明對Foc譜系VI、TR4和STR4的qPCR檢測具有重復性和可重復性,其中,LOQ值為10-4~10-3ng/μL,LOD為10-5~10-4ng/μL。
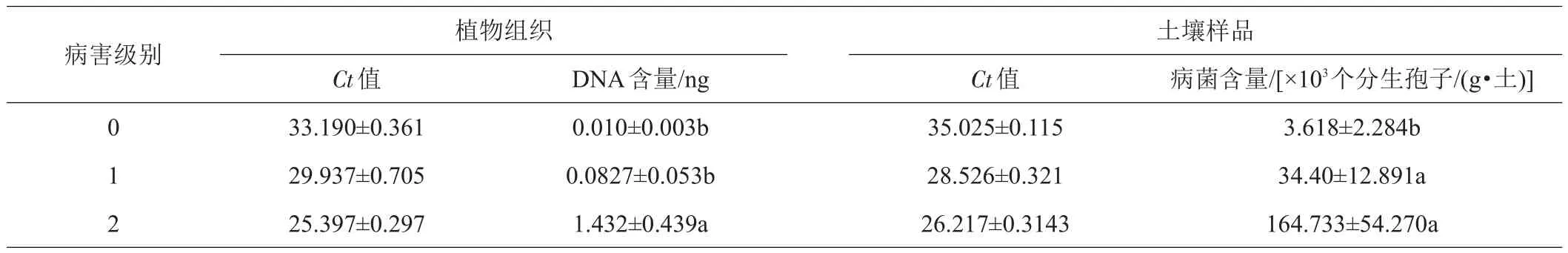
表1 不同病害級別荷花植株中荷花腐敗病菌DNA的含量及土壤中荷花腐敗病菌的含量
對各種發病級別的荷花地下莖組織及土壤中荷花腐敗病菌的含量與發病程度進行相關分析,結果表明,荷花植株中荷花腐敗病菌的數量與其發病的程度呈顯著的正相關,而土壤中荷花腐敗病菌的數量與發病的程度相關性低(無顯著的相關性),但荷花腐敗病菌的數量還是隨著病害發生程度的增加而增加。當然,本研究也存在著一些不足,比如病害的分級偏少、實驗樣本的處理以及qPCR反應優化問題[26]等。因此,在后續的研究中,還需要建立明確的荷花腐敗病病害分級系統,進一步分析土壤或植株中病原菌數量與病害發生程度的關系,從而指導荷花腐敗病的防控。綜上所述,本研究建立的qPCR定量檢測方法是研究此類土傳病害強有力的檢測手段。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
