鈣離子穩態的調控在糖尿病相關心房顫動中的作用
2021-11-13 07:32:10高婧晗劉飛楊曉蕾夏云龍
心血管病學進展 2021年10期
關鍵詞:糖尿病
高婧晗 劉飛 楊曉蕾 夏云龍
(大連醫科大學附屬第一醫院,遼寧 大連 116011)
糖尿病是最常見的慢性病并且是心血管疾病的主要危險因素[1],可明確增加心房顫動(房顫)的發生風險,研究表明糖尿病患者比非糖尿病患者發生房顫的風險高約40%[2],并且糖尿病與房顫患者癥狀的嚴重程度和遠期生存質量密切相關[1]。同時,糖尿病患者合并房顫發生腦卒中和心力衰竭等并發癥的風險顯著高于未罹患糖尿病的患者[3]。另一方面,糖尿病合并房顫的患者大約有1/3預后不良,且死亡率明顯高于不合并糖尿病的患者[4]。
糖尿病誘發房顫的機制包括心房結構重構及心房電重構,且兩者之間相輔相成,相互影響[5]。心房結構重構的主要表現包括心房擴大和心房間質纖維化[6]。其中,心房間質纖維化可導致心房傳導延遲和折返環路的形成[7]。另一方面,心房結構重構的發生往往伴隨著電學特征的改變[8]。主要表現為快速心房搏動引起心房肌動作電位時程(action potential duration,APD)及心房有效不應期(atrial effective refractory period,AERP)的縮短、AERP頻率適應性下降等[9],進而導致一系列心律失常的發生。研究表明,細胞內外鈣離子穩態的失衡是導致房顫發生和發展的重要原因[10]。近些年越來越多的研究表明,鈣離子穩態的異常在糖尿病相關房顫的發生和發展中起關鍵作用[11]。現綜述鈣離子穩態的調節失衡在糖尿病患者發生房顫的發病機制方面的相關進展(見圖1)。
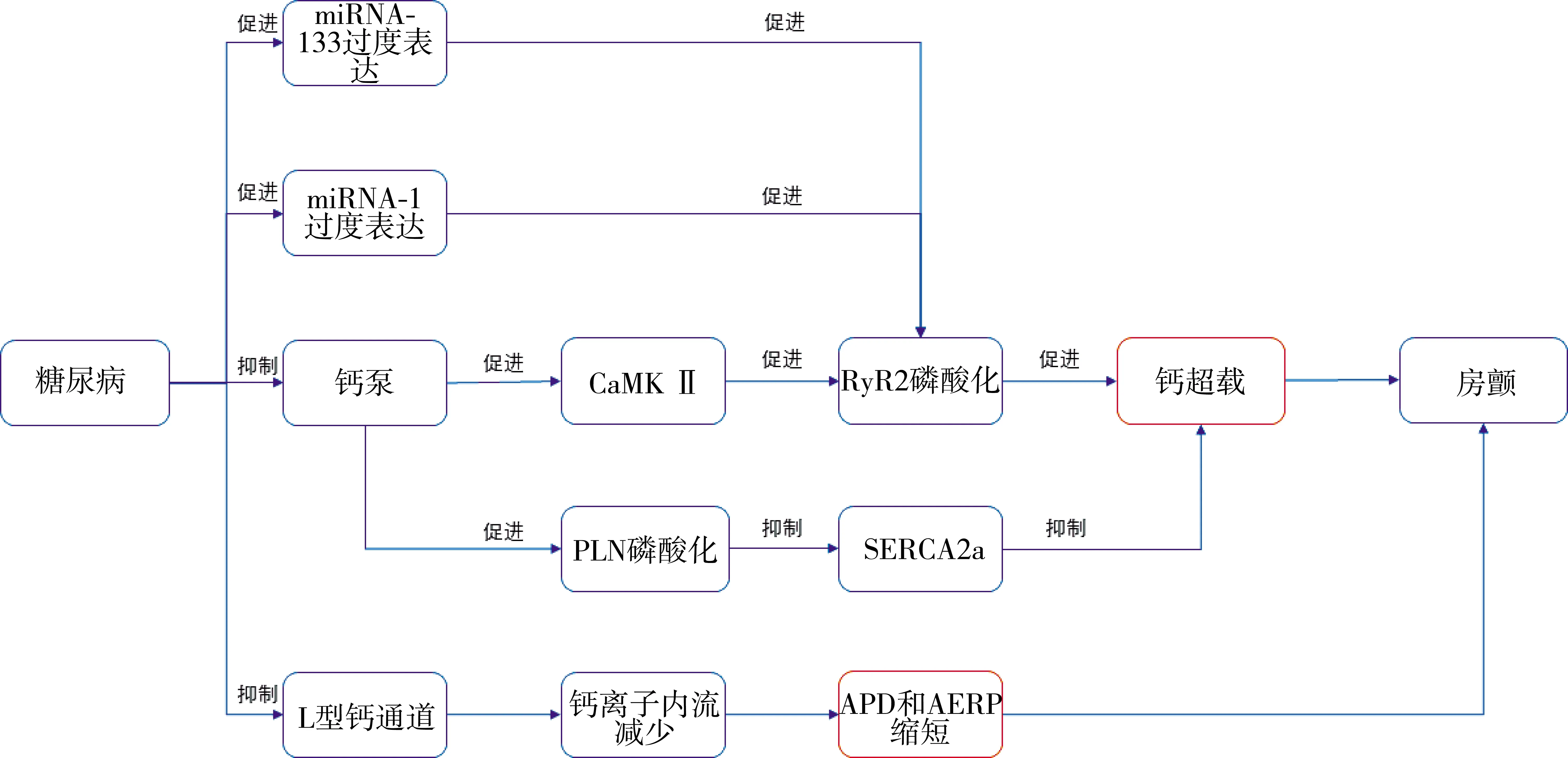
注:RyR2:雷諾丁受體2;PLN:受磷蛋白;SERCA2a:肌漿網鈣離子ATP酶2a。圖1 鈣離子在調控糖尿病相關房顫中的作用機制
1 鈣調蛋白激酶Ⅱ在糖尿病患者中誘發房顫的發生機制
鈣離子主要來源于肌漿網鈣離子的釋放和經細胞膜L型鈣通道對細胞外鈣離子的攝取[10]。鈣離子濃度主要受鈣調蛋白信號通路的調節,研究表明,高血糖可引發心肌細胞內鈣泵功能的降低,導致心肌細胞內鈉鈣交換增加和鈣通道的激活,進而使得細胞內鈣離子濃度升高,從而增加細胞內鈣離子和鈣調蛋白(calmodulin,CaM)結合的概率,激活CaM和Ca2+-鈣調蛋白依賴性蛋白激酶(Ca2+/calmodulin-dependent protein kinase,CaMK),導致鈣調蛋白信號通路的過度激活[12]。同時研究表明,急性高血糖可引起N-乙酰氨基葡萄糖對CaMKⅡ進行共價修飾,從而自動激活CaMKⅡ,進一步促進鈣調蛋白信號通路的激活[13]。因此,CaMKⅡ在糖尿病患者發生房顫的作用機制中起重要作用。
1.1 CaMKⅡ對雷諾丁受體的調控作用
鈣離子與CaMKⅡ結合后,可激活肌漿網上的雷諾丁受體(ryanodine receptor,RyR),從而促進鈣離子進一步從肌漿網上釋放,引發鈣誘導鈣釋放的發生[14]。研究表明,CaMKⅡ的過度激活,增加了RyR的開放頻率,從而使得房顫中肌漿網自發性鈣釋放增加,引起細胞內鈣超載[15]。有實驗證明,在糖尿病大鼠的心肌細胞中可觀察到RyR2激活增強導致的自發性的鈣釋放[16]。同時高糖飲食的大鼠,CaMKⅡ誘導的RyR2磷酸化程度相比于正常飲食的大鼠明顯增加,而RyR2磷酸化會增加肌漿網鈣離子泄漏,使得細胞內鈣離子濃度增高,促進房顫的發生[17]。因此,CaMKⅡ抑制劑成為糖尿病患者治療房顫等心律失常的潛在治療策略[18]。
1.2 CaMKⅡ對肌漿網鈣離子ATP酶2a的調控作用
研究表明,CaMKⅡ通過特異性調控受磷蛋白(phospholamban,PLN)磷酸化進而影響肌漿網鈣離子ATP酶2a(sarcoplasmic reticulum Ca2+ATPase,SERCA2a)的活性,因此,PLN是CaMKⅡ的特異性磷酸化位點[18]。PLN可與SERCA2a相互作用并抑制其活性,從而抑制鈣離子通過SERCA2a進入肌漿網,同時研究表明PLN的磷酸化狀態會影響SERCA2a的活性[19]。實驗表明磷酸化的PLN通過與心肌細胞內鈣離子結合,進一步觸發了自發性鈣離子釋放,促進細胞內鈣離子濃度升高,引發鈣超載,從而引起房顫等心律失常的發生[18]。研究表明,SERCA2a在糖尿病小鼠模型的心肌內表達明顯降低[20],這與肌漿網對鈣離子的攝取減少密切相關[21]。同時,也有研究表明在糖尿病心肌細胞中,總體上SERCA2a水解ATP和轉運鈣離子的能力相比于正常心肌細胞顯著降低[22],從而引起細胞內鈣離子濃度異常,引發房顫等一系列心律失常。
2 L型鈣通道在糖尿病心肌病中對鈣穩態的調控
心房電重構主要涉及L型鈣通道功能改變以及電壓依賴性L型鈣通道蛋白的失活[10]。研究表明,糖尿病導致的病理損傷可引起離子通道發生改變[11]。有實驗研究表明,高糖狀態可引起L型鈣通道表達減少[23],引起細胞內鈣離子濃度異常,使得APD縮短,導致異常的心房電沖動和糖尿病相關性房顫的發生。另一方面,糖尿病大鼠心肌細胞在舒張期肌質網鈣離子儲存減少[23],而房顫發作時出現的快速心房收縮,使得肌漿網鈣離子攝取異常,進一步促進了細胞內鈣超載的發生[24]。由于細胞內鈣離子的升高,心房肌細胞進一步下調L型鈣通道蛋白的表達[25],引起鈣電流密度進一步降低[23],導致APD和AERP縮短,進而導致心房傳導速度降低和折返激動得以維持,促進了房顫的發生和發展[10,26]。此外,房顫發作時不僅下調L型鈣通道蛋白的表達,還可通過蛋白的去磷酸化、促進通道蛋白降解等途徑來降低L型鈣通道的功能和活性,進一步抑制了鈣離子內流[27]。
3 小分子核糖核酸在糖尿病心肌病中對鈣穩態的調控作用
小分子核糖核酸(microRNA,miRNA)參與了包括細胞凋亡、細胞增殖等在內的生命活動。近些年,研究表明miRNA的表達失調可導致多種離子通道表達異常,最終導致心房重構[10]。而糖尿病對于miRNA的表達具有重要調控作用。miRNA通過與Argonaute蛋白家族結合形成RNA誘導沉默復合體[28],使得目標mRNA的翻譯水平下降,從而抑制目的基因的表達。
miRNA-1是心臟中表達量最高的miRNA。miRNA-1的過度表達可抑制細胞L型鈣通道的表達,促進房顫的發生[29]。研究表明,糖尿病的細胞高糖狀態導致的細胞毒性作用,可引起心肌細胞miRNA-1過度表達[30]。miRNA-1過度表達會促進RyR2的磷酸化,使得細胞內鈣離子釋放增加,引起細胞內鈣離子濃度異常,引起房顫等心律失常的發生[28]。同時研究表明,在糖尿病兔心臟中miRNA-133水平顯著上調[30],miRNA-133過度表達可抑制蛋白磷酸酶的活性,導致RyR2過度磷酸化,引起肌漿網鈣離子釋放增加和鈣超載的發生,增加發生房顫等心律失常的風險[29]。另一方面,miRNA-133的過度表達能降低心肌細胞葡萄糖轉運蛋白4水平,降低胰島素誘導的葡萄糖水平,導致心肌細胞中葡萄糖攝取減少[30]。故而miRNA-1和miRNA-133的過度表達,可引起RyR2過度磷酸化,進而細胞內鈣釋放增加,導致房顫的發生與發展[31]。
因此miRNA可能參與了糖尿病患者房顫誘導的心房電重構,引起細胞內鈣穩態異常,導致持續性房顫的發生[32]。故而針對miRNA的特異性治療成為了抗心律失常治療的潛在靶點。有研究證明,在轉基因小鼠中磷酸化向去磷酸化的轉化可能具有抗心律失常的作用,這些結果表明,心律失常事件的增加可能是由于miRNA-1和miRNA-133介導的RyR2磷酸化的結果,這為建立特異性miRNA-1和miRNA-133治療靶點提供了理論基礎[31]。
4 展望
糖尿病是目前最常見的慢性疾病之一,可明顯增加房顫的發生風險,并且兩者常常合并存在,嚴重威脅人類的生命健康。糖尿病不僅會增加房顫患者的死亡率,并且會降低患者遠期生存率,降低長期生存質量。糖尿病通過調控離子通道異常和CaMKⅡ的過度激活,引起細胞內鈣離子穩態異常,進而導致房顫的發生。另一方面miRNA介導的鈣離子通道表達異常和CaMKⅡ介導的SERCA2a表達的減少,也在細胞內鈣離子穩態失衡中發揮了重要作用。但關于具體的分子調控機制以及潛在的治療靶點仍需進一步的研究。
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年3期)2021-08-22 06:49:56
中老年保健(2021年11期)2021-08-22 03:15:16
中國生殖健康(2020年2期)2021-01-18 02:51:44
中國生殖健康(2018年2期)2018-11-06 07:11:04
基層中醫藥(2018年2期)2018-05-31 08:45:04