成年型球形細胞腦白質營養不良1例
2021-10-21 12:08:24馮雪丹于莎莎張玉靖祖煜高煜陳劍華馬紅梅王向波
疑難病雜志 2021年10期
馮雪丹,于莎莎,張玉靖,祖煜,高煜,陳劍華,馬紅梅,王向波
患者,女,60 歲,主因“進行性雙下肢無力 2 年余” 于2017年11月入院。患者2年前出現雙下肢僵硬無力、行走不穩,伴頭部不自主抖動。18個月出現四肢末端麻木及前胸部束帶感。雙下肢無力逐漸進展,6個月前不能站立行走,抬離床面困難,伴小便失禁、言語笨拙。多次就診于各大醫院,行頭部及頸胸椎MR 平掃示腦內、頸、胸髓內多發異常信號,腦萎縮。考慮 “多發性硬化”,給予改善循環、營養神經等藥物,并多次行激素、丙種球蛋白治療無明顯好轉。既往糖尿病史10余年。查體:雙上肢肌力Ⅴ級,雙下肢肌力Ⅲ級,雙下肢肌張力增高,腱反射活躍,雙側Babinski征、Chaddock 征陽性。腦脊液IgA 1.69 mg/dl(正常參考值0~0.2 mg/dl),IgG 11.9 mg/dl(正常參考值0.48~5.86 mg/dl),腦脊液IgG寡克隆區帶(OB)及特異性IgG 寡克隆區帶(SOB)均陽性。頭顱MR平掃顯示雙側額葉、側腦室旁、基底節區、顳葉海馬及腦干異常密度信號灶,雙側側腦室對稱性擴大,腦溝、腦裂增寬;胸椎MR平掃可見脊髓內線狀長T2密度信號灶,軸位掃描見病灶于脊髓背側(圖1)。第二代高通量測序結果顯示,受檢者(患者及其兒子)β-半乳糖腦苷脂酶(GALC)基因 c.1418G>A的雜合核苷酸變異(圖2)。代謝腦病4項中GALC活性為4.2 nmol·mg-1·17 h-1(正常參考值18~75 nmol·mg-1·17 h-1)。診斷為成年型球形細胞腦白質營養不良(Krabbe)病,給予B族維生素營養神經、巴氯芬緩解肢體痙攣狀態后好轉出院。患者出院后繼續長期口服上述藥物,每年均靜脈輸注丙種球蛋白,臨床癥狀無明顯進展。
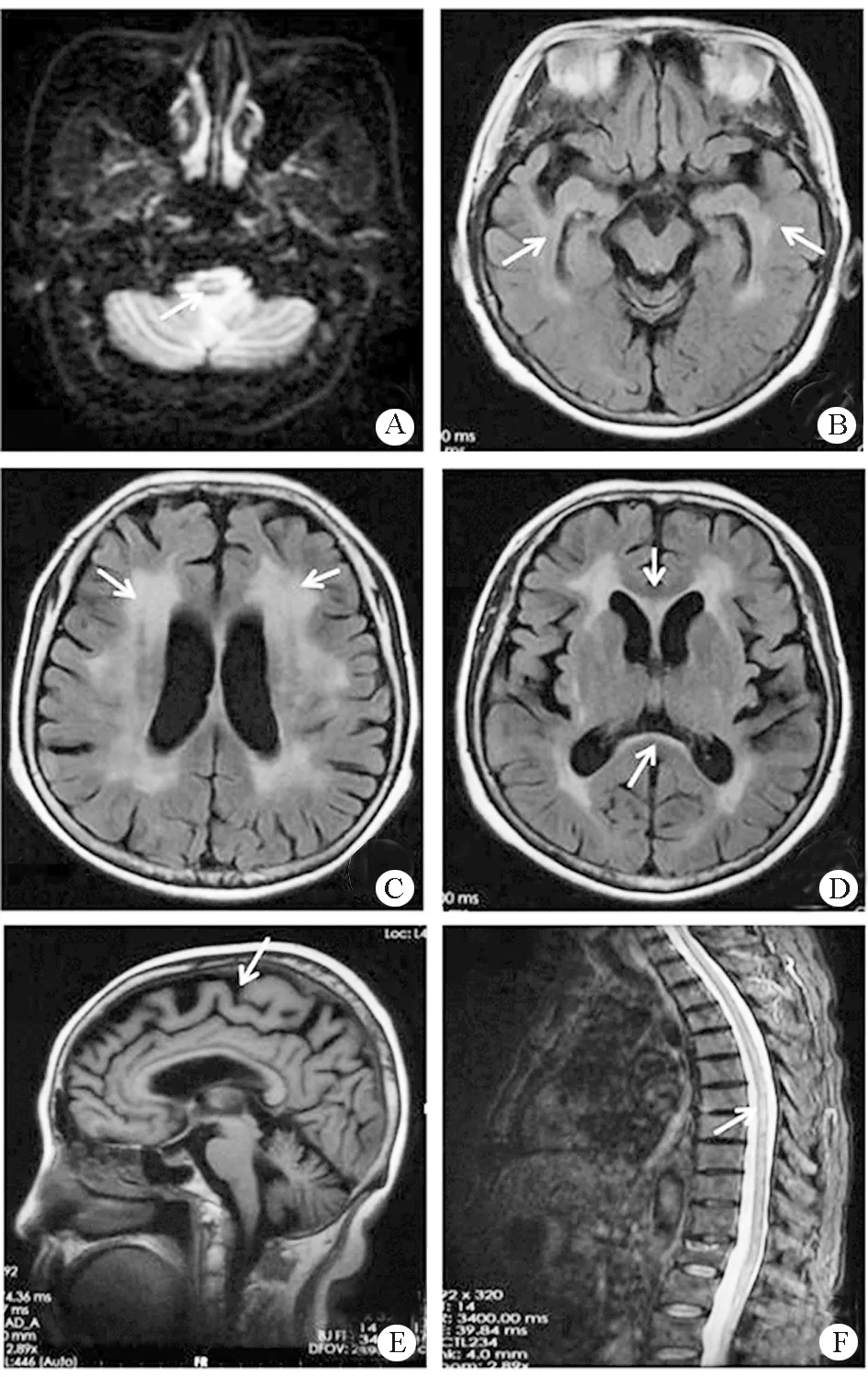
注:A~D.腦干、雙側海馬、胼胝體、側腦室旁異常密度信號灶;E.彌漫性腦萎縮;F.胸髓內見線狀長T2密度信號
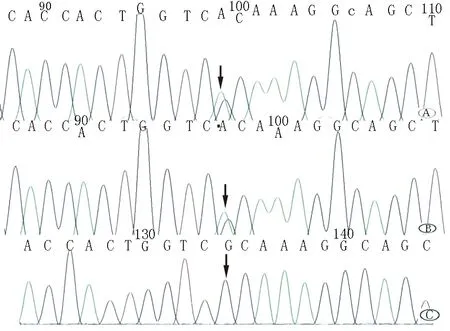
注:患者(A)及其子(B)c.1418G>A雜合變異,紅色箭頭所指為變異位點,患者丈夫(C)未發現變異
討 論球形細胞腦白質營養不良(globoid cell leukodystrophy,GLD)又稱Krabbe病,是由于GALC基因缺陷導致半乳糖腦苷脂蓄積于神經系統而引發的疾病,確診的主要依據是GALC活性缺乏和基因學檢測。該病發病率約為1 /10萬,主要發生于嬰幼兒,超過20歲的成年型不足5%[1-2]。本例為國內報道起病年齡最大的患者。成年型Krebbe病較早發型癥狀輕、進展緩慢,主要表現為痙攣性截癱、步態障礙、精神障礙、視聽障礙、語言障礙、周圍神經病變等[3-4],本例符合成年型Krabbe病臨床表現。因為本病可表現為不對稱的肢體無力和行走困難,所以與多發性硬化難以鑒別[4-5]。
Krabbe病的典型影像學特征是皮質脊髓束或腦室周圍白質對稱性異常信號,少數僅顯示輕度腦萎縮[3,6]。本例患者MR提示病變累及雙側額葉、腦室旁、基底節區及胼胝體壓部,以及雙側顳葉海馬、腦干和脊髓這些少見部位。脊髓內散在片條狀異常信號與多發性硬化相似,兩者鑒別點在于多發性硬化多不具有對稱性和沿皮質脊髓束分布的特點。此外,90%Krabbe病患者電生理檢查可發現周圍神經受損,部分為亞臨床病變[7],而本例MR及肌電圖無周圍神經受損的證據。Krabbe病患者腦脊液檢查大多正常,或有輕度蛋白升高。本例與Tomás等[5]報道的病例相似,腦脊液OB、SOB陽性,IgG指數和24h IgG合成率異常而在血清中正常,提示腦脊液存在體液免疫反應。目前認為Krabbe病的機體免疫反應激活先于臨床癥狀和白質病理性改變[8]。本例患者出院后定期輸注丙種球蛋白,隨訪3年病情穩定。免疫治療是否可使Krabbe病患者獲益需進一步證實。Krabbe病尚無特異性根治措施,在癥狀出現之前行造血干細胞移植(HSCT)是唯一證實有效的治療方法,基因治療和酶替代療法仍處于實驗階段。所以,對于Krabbe病家系通過產前診斷避免基因缺陷患兒的出生更具意義。
Krabbe病由GALC基因突變引起,該基因位于14q31號染色體,編碼GALC,對鞘糖脂的分解代謝發揮至關重要的作用。人類基因突變數據庫中已經編錄了至少237個導致Krabbe病的突變基因[2]。在本例患者及其兒子GALC基因中發現c.1418G>A的雜合核苷酸變異為錯義變異,該變異導致第473號氨基酸由精氨酸變為組氨酸(p.Arg473His),進而導致蛋白質功能受到影響。患者兒子37歲尚未發病或為延遲顯性,由于缺少受檢者父母檢驗樣本,所以未進行父母來源驗證工作。c.1418G>A變異為新的致病突變,在100例正常人、基因多態性數據庫、人類基因突變數據庫中均未檢出該突變,國內外文獻也未見相關報道。新突變基因c.1418G>A的發現拓展了Krabbe病的基因突變譜,或為成年型Krabbe病的特殊變異。
本例患者臨床表現、實驗室及影像學表現符合MacDonald原發進展型多發性硬化診斷標準,多次誤診為“多發性硬化”,顱腦影像學顯示病變沿皮質脊髓束分布的特點、酶學及基因學檢測是2種疾病鑒別診斷的關鍵點。
