HPLC法測定太子參藥材中多糖水解產物單糖的含量
2021-10-08 07:55:22王慧娟彭禮軍
中國民族民間醫藥 2021年15期
王慧娟 喬 楊 張 敏 彭禮軍
1.西南藥食兩用資源開發利用技術國家地方聯合工程研究中心,貴州 貴陽 550025;2.黔西南民族職業技術學院,貴州 興義 562400
太子參為石竹科植物孩兒參Pseudostellariaheterophylla(Miq.) Pax ex Pax et Hoffm. 的干燥塊根,中醫臨床常用補益藥[1]。近年來,人們對太子參化學成分進行了比較系統的研究[2]。研究[3-6]表明,多糖是太子參的重要活性成份,具有抗應激、抗氧化和調節機體免疫的作用,對脂多糖(LPS)誘導的大鼠心肌細胞損傷和大鼠急性心肌梗死誘發的心肺損傷有一定的保護作用。
太子參多糖主要由葡萄糖、半乳糖、木糖、鼠李糖等組成[7]。本研究在此基礎上,建立柱前衍生化-HPLC法測定太子參多糖中鼠李糖、葡萄糖、半乳糖和木糖的含量,為太子參藥材質控標準的完善提供基礎。
1 材料
Agilent 1260高效液相色譜儀(美國安捷倫公司),FA2004型分析天平(上海良平儀器儀表有限公司),GZX-9146MBE數顯鼓風干燥箱(上海博迅實業有限公司醫療設備廠),恒溫水浴鍋(金壇市大地自動化儀器廠)。
太子參藥材(貴州百靈制藥有限公司);1-苯基-3-甲基-5-吡唑啉酮(PMP)(天津科密歐化學試劑有限公司);葡萄糖(成都植標化純生物技術有限公司,含量≥98.0%);半乳糖(solarbio公司,含量≥99.0%);鼠李糖(上海源葉生物科技有限公司,含量≥98.0%)、木糖(上海源葉生物科技有限公司,含量≥99.0%);醋酸銨、氯仿、氫氧化鈉、甲醇、濃鹽酸、三氟乙酸均為分析純,乙腈為色譜純。
2 方法與結果
2.1 液相色譜條件優化
2.1.1 混合單糖對照品衍生化供試液制備 對照品溶液的制備:精密稱取葡萄糖、木糖、鼠李糖、半乳糖各100mg,加水溶解,定容至100 mL。
取對照品溶液1 mL,依次加入PMP甲醇溶液(0.5 moL·L-1)和NaOH溶液(0.2 moL·L-1)各0.5 mL,混勻,70 ℃水浴100 min,冷卻,加入鹽酸溶液(0.2 mol·L-1)中和,混勻,三氯甲烷洗滌3次,每次2 mL,取水層,離心后上清液用微孔濾膜過濾,取續濾液,即得。
2.1.2 色譜柱選擇 分別采用waters C18(4.6 mm×250 mm,5 μm),Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 μm)和Agilent Eclipse XDB-C18(4.6 mm×250 mm,5 μm)三種色譜柱,考察色譜柱對混合對照品衍生化供試液色譜峰的影響。結果表明,Agilent Eclipse XDB-C18色譜柱能對各衍生化組分進行很好地分離。具體如圖1所示。

1.waters C18 2.Agilent ZORBAX SB-C18 3.Agilent Eclipse XDB-C18
2.1.3 柱溫篩選 采用Agilent Eclipse XDB-C18色譜柱,設定柱溫為25 ℃,30 ℃和35 ℃。結果發現,柱溫越高,保留時間越短,出峰越快。為節約時間,選取柱溫為35 ℃。
2.1.4 流動相考察 采用Agilent Eclipse XDB-C18色譜柱,設定流動相為乙腈-水、乙腈-乙酸銨溶液(0.01 moL·L-1)、乙腈-乙酸銨溶液(0.02 moL·L-1)和乙腈-乙酸銨溶液(0.05 moL·L-1)。結果發現,乙酸銨對峰形及分離度有改善作用,濃度越高,峰形對稱性越好,但對設備損傷較大。綜合考慮,流動相選取乙腈-乙酸銨溶液(0.02 moL·L-1)。
2.2 太子參多糖水解方法篩選
2.2.1 水解用酸的考察 取6份太子參多糖(自制)各10 mg,加水1.0 mL溶解,分別加入3、5、7 moL·L-1鹽酸和1、3、5 moL·L-1三氟乙酸溶液各1.0 mL,混勻,110 ℃水解1 h,放冷,NaOH溶液中和,得太子參多糖水解液。
取上述6份多糖水解液各1.0 mL,照“2.1.1”項下處理后測定,其結果見表1。

表1 多糖酸水解后各單糖衍生物的峰面積
實驗表明,不同水解條件下4種單糖峰面積變化趨勢并不相同。綜合考慮,水解用酸選擇3 moL·L-1鹽酸溶液。
2.2.3 水解溫度考察 取3份太子參多糖各10 mg,加水1.0 mL溶解,分別加入鹽酸(3 mol·L-1)1.0 mL,混勻,分別在100 ℃、110 ℃、120 ℃水解1 h,放冷, NaOH溶液中和,得太子參多糖水解液。
取上述多糖水解液各1.0 mL,照“2.1.1”項下處理后測定,根據實驗測定結果,水解溫度確定為110 ℃,其結果見表2。

表2 不同溫度水解多糖后各單糖衍生物的峰面積
2.2.4 水解時間考察 取3份太子參多糖各10 mg,加水1.0 mL溶解,加入鹽酸(3 moL·L-1)1.0 mL,混勻,于110 ℃分別水解30,60 和90 min,放冷, NaOH溶液中和,得太子參多糖水解液。
取上述多糖水解液各1.0 mL,照“2.1.1”項下處理后測定,根據實驗測定結果,水解時間確定為30 min,具體見表3。

表3 不同水解時間對單糖衍生物峰面積的影響
2.3 衍生化方法的篩選
2.3.1 衍生化溫度的篩選 取太子參多糖水解液3份,每份1.0 mL,置10 mL具塞試管中,依次加入PMP甲醇溶液(0.5 moL·L-1)和NaOH溶液(0.2 moL·L-1)各0.5 mL,混勻,分別于40 ℃,70 ℃,95 ℃水浴100 min,處理后進樣測定。綜合考慮各單糖峰面積,將衍生化溫度確定為70 ℃,具體見表4。

表4 衍生化反應溫度對單糖峰面積的影響
2.3.2 衍生化時間的篩選 取太子參多糖水解液3份,每份各1.0 mL,依次加入PMP甲醇溶液(0.5 moL·L-1)和NaOH溶液(0.2 moL·L-1)各0.5 mL,混勻,于70 ℃水浴分別加熱30,60和100 min,處理后進樣測定。根據各單糖峰面積的大小確定衍生化時間為100 min,具體見表5。

表5 衍生化時間對單糖峰面積的影響
2.4 太子參藥材中多糖水解產物單糖的含量測定
2.4.1 色譜條件 色譜柱:Agilent Eclipse XDB-C18(4.6 mm×250 mm,5 μm);流動相:乙腈-0.02 moL·L-1的乙酸銨溶液(19∶81);檢測波長:250 nm;流速:1.0 mL·min-1;柱溫:35℃;進樣量:10 μL。
2.4.2 供試品溶液的制備及測定 供試品溶液的制備 精密稱取太子參粉末0.2 g,置索氏提取器中,用80 mL乙醇回流提取1 h。過濾,濾渣揮干乙醇后,加水40 mL回流提取4 h,趁熱抽濾,再加水30 mL回流提取2 h,抽濾,合并濾液,定容至100 mL。精密吸取1 mL定容至10 mL,作為供試品溶液。
測定法:取太子參供試品溶液1 mL,加鹽酸溶液(3.0 moL·L-1)1 mL,混勻,110 ℃水解30 min,放冷,NaOH溶液調pH值至中性。取1 mL,依次加入PMP甲醇溶液(0.5 mol·L-1)和NaOH溶液(0.2 moL·L-1)各0.5 mL,混勻,70 ℃水浴加熱100 min,冷卻,鹽酸溶液(0.2 moL·L-1)中和后定容至5 mL,混勻,三氯甲烷洗滌3次,每次2 mL,取水層,離心后將上清液過濾,取續濾液,測定,即得。
2.4.3 系統適用性試驗 分別吸取混合對照品液、供試品溶液進行測定,記錄色譜圖(見圖2)。從圖中可見,供試品溶液和對照品溶液中鼠李糖、葡萄糖、半乳糖、木糖的保留時間一致,且供試品溶液中各待測峰的分離度符合要求。

A.混合單糖對照品溶液;B.供試品溶液
2.4.4 線性關系考察 取四種單糖濃度均為1 mg·mL-1的混合單糖對照品溶液,衍生化處理后測定,記錄色譜圖。以進樣量x(μg)為橫坐標,峰面積A為縱坐標,計算回歸得到四種單糖的標準曲線,見表6。

表6 四種單糖衍生物的標準曲線
2.4.5 精密度試驗 取太子參藥材(1號)0.2 g,按“2.4.2”項下制備供試液,連續進樣6次,測定各單糖的峰面積,結果見表7。RSD均小于2%,表明儀器精密度良好。
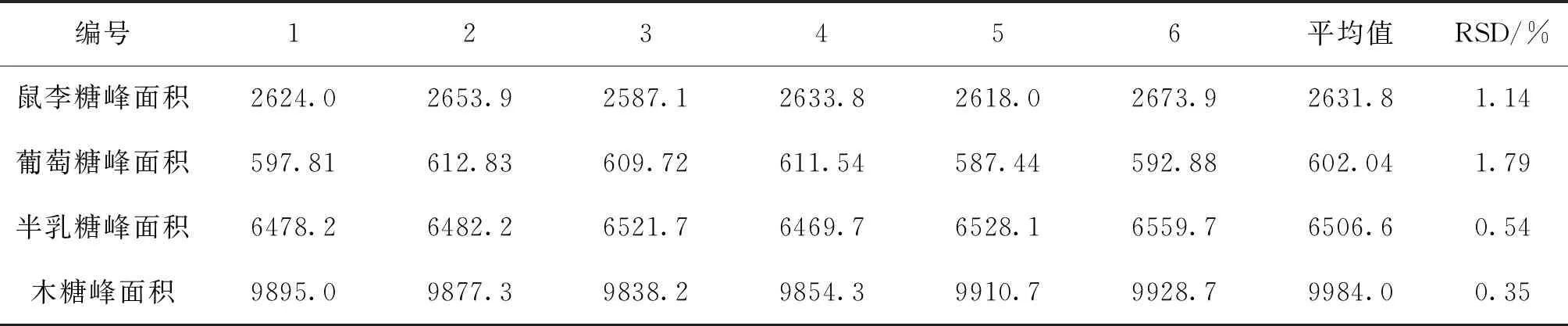
表7 精密度試驗結果
2.4.6 重復性試驗 取太子參藥材(1號)0.2 g,按“2.4.2”項下方法制備供試品溶液6份,分別進樣,測定峰面積,結果見表8。

表8 重復性試驗
2.4.7 穩定性試驗 取太子參藥材(1號)0.2 g,按“2.4.2”項下制備供試品溶液,分別于衍生化后0、2、4、8、12、24、48 h進樣測定,結果列入表9。RSD均小于2%,說明供試液衍生化后在48 h內穩定性良好。

表9 穩定性試驗
2.4.8 加樣回收率試驗 精密稱取6份已測定含量的樣品(樣品編號1號)各0.1g,置索氏提取器中,加乙醇回流提取1 h,濾渣揮干乙醇,再精密加入適量的鼠李糖、葡萄糖、半乳糖、木糖對照品,按照 “2.4.2”項下處理后測定,計算回收率,結果見表10~13。

表10 加樣回收率試驗(鼠李糖)
2.4.9 樣品測定 取不同產地的太子參藥材樣品,分別制備供試品溶液后測定,結果見表14。

表11 加樣回收率試驗(葡萄糖)

表12 加樣回收率試驗(半乳糖)

表13 加樣回收率試驗(木糖)

表14 太子參藥材中各單糖含量測定結果
結果表明:各批次太子參的單糖含量懸殊不是很大,且皆呈現出同樣趨勢:其含量木糖﹥半乳糖﹥鼠李糖﹥葡萄糖。各批次藥材中木糖和半乳糖含量均在10%以上,葡萄糖含量較低,大多在5%以下。
3 討論
本文采用柱前衍生化-HPLC法對太子參藥材中的鼠李糖、葡萄糖、半乳糖以及木糖進行測定,分別對液相色譜條件、多糖水解和衍生化條件進行了優化,比較了不同條件下單糖衍生物的峰形、分離度、峰面積大小變化等,最終確定了太子參藥材中多糖水解產物單糖的含量測定方法。
對所建方法進行方法學驗證,結果發現4種單糖標準曲線的線性均較好(回歸系數0.9999),精密度、重復性及穩定性試驗的RSD均小于3%。在加樣回收率試驗中,鼠李糖、半乳糖和木糖的平均回收率在95%~105%之間,且RSD﹤3%,符合要求。但葡萄糖的回收率偏低,平均值為86.04%,這可能與太子參藥材中葡萄糖含量較低且樣品前處理過程比較復雜有關。因此,在后續的太子參質量標準研究工作中,可優先考慮將含量較高的鼠李糖、半乳糖和木糖納入質控標準。
本文的研究工作,可為今后太子參藥材質量標準的補充完善提供研究基礎及數據支撐,對太子參的質量控制具有一定的價值和意義。
