胰島素信號通路在運動改善阿爾茨海默病中的作用機制
2021-09-29 01:23:50任鵬飛
體育科研 2021年5期
任鵬飛
阿爾茨海默病(Alzheimer's Disease,AD)是一種神經系統退行性疾病,是老年癡呆中最常見的一種疾病。2017年,全球約有4 400萬AD患者,到2050年,全球AD患者可能達到1.15億[1]。AD主要臨床表現為認知功能障礙和記憶力衰退,其病理學特征為β-淀粉樣蛋白(β-amyloid,Aβ)沉積、Tau蛋白過度磷酸化、神經元丟失和神經元內顆粒空泡變性,此外還包括糖代謝異常以及氧化應激增加等[2]。人體研究發現,外周胰島素抵抗會增加AD發病機率,且大部分AD患者存在胰島素抵抗和胰島素分泌缺陷,因而AD也被認為是一種“腦型糖尿病”[3]。此外,動物實驗也表明,AD轉基因小鼠腦內存在胰島素抵抗[4]。而抑制AD腦內胰島素抵抗可以顯著改善AD病理表現[5]。胰島素信號通路受損是出現腦部胰島素抵抗的重要原因,因而改善AD腦內胰島素信號通路受損,可能是緩解AD的有效途徑。其中,磷脂酰肌醇-3-激酶/絲蘇氨酸激酶(Phosphatidylinositol-3-Kinases/Serine Threonine Kinase,PI3K/AKT)、絲裂原活化蛋白酶(Mitogen-Activated Protein Kinase,MAPK)、Wnt通路是3條主要的胰島素信號通路[6]。
已有研究證明,運動可以減少AD腦內Aβ沉積,抑制Tau蛋白過度磷酸化,改善細胞自噬水平,顯著提高學習記憶能力,但運動改善AD的作用機制仍不明晰。現有文獻表明,運動可能通過激活PI3K/AKT、MAPK和Wnt信號通路,改善AD病理。因此,本文將從上述3條胰島素信號通路切入,進行研究綜述。
1 PI3K/AKT信號通路與AD
PI3K/AKT信號通路受胰島素、神經生長因子等調控,參與細胞增殖、分化、存活等諸多生物學功能。激活后的PI3K和AKT的PH結構域結合,改變構象并發生位移,磷酸化AKT的Ser473和Thr308位點從而激活AKT,活化的AKT作用于糖原合成酶3(Glycogen Synthase Kinase3,GSK3)和哺乳動物雷帕霉素靶蛋白(Mammalian Target of Rapamycin,mTOR)等下游因子,調節細胞的生長和凋亡[7]。目前研究已經證實,PI3K/AKT信號通路可能通過調控Tau蛋白、Aβ、神經炎癥和自噬,調控AD病理特征。
1.1 運動激活PI3K/AKT減少Aβ沉積
Aβ是由β-淀粉樣前體蛋白(β-Amyloid Protein Precursor,APP)被β-分解酶和γ-分解酶水解后生成的。正常情況下,腦內Aβ的生成與降解存在動態平衡,在AD中這種平衡被打破,腦內Aβ大量聚集。研究顯示,PI3K/AKT信號活性降低時,Aβ出現過度沉積[8]。中藥清心開竅方可激活APP/PS1轉基因小鼠海馬組織PI3K/AKT信號通路,降低Aβ水平[9]。且激活PI3K/AKT信號通路可通過降低β-分泌酶活性減少Aβ含量[10]。進一步研究證實,降低PI3K/AKT信號通路活性可以增加糖原合成激酶-3α(Glycogen Synthase Kinase-3α,GSK-3α)磷酸化,提高β-分泌酶活性,促進Aβ40和Aβ42生成。同時,Aβ40和Aβ42可通過增加Ca2+進入神經元,激活GSK-3β和細胞周期素依賴蛋白激酶5(Cyclin-Dependent Kinase5,CDK5),進一步阻斷PI3K/AKT通路活性,最終導致Aβ過度沉積和AD認知功能障礙[11]。提示增加PI3K/AKT信號通路活性可以通過降低GSK-3α磷酸化水平,降低β-分泌酶,抑制AD腦內Aβ生成。
有研究報道,5個月跑臺運動降低了APP/PS1小鼠海馬組織中GSK-3α的Ser21位點磷酸化水平,抑制了Aβ沉積[12]。6周跑臺運動可以提高糖尿病鼠海馬內AKT表達,降低GSK-3α磷酸化,減少Aβ沉積,改善認知功能[13]。提示運動可能通過改善PI3K/AKT信號通路活性,降低GSK-3α磷酸化水平,減少Aβ沉積。另有研究發現,8周有氧運動激活了小鼠皮質和海馬組織中PI3K/AKT信號通路,降低了β-分泌酶水平,減少了Aβ42表達,但APP蛋白含量并沒有發生顯著改變[14]。提示有氧運動通過激活PI3K/AKT信號通路,抑制β-分泌酶產生,使APP蛋白向非Aβ途徑轉移,產生可溶的小分子蛋白,減少Aβ生成。以上研究說明,運動可以激活PI3K/AKT信號通路,降低GSK-3α磷酸化水平,減少β-分泌酶含量,從而抑制Aβ生成,改善AD認知能力。
1.2 運動激活PI3K/AKT改善Tau蛋白過度磷酸化
Tau蛋白異常磷酸化是AD主要病理特征之一。對AD患者尸檢發現,顳葉皮層中過度磷酸化的Tau蛋白顯著增多,同時伴隨有磷酸化AKT水平顯著降低[15]。Schubert等[16]也指出,抑制PI3K/AKT信號通路會導致Tau蛋白過度磷酸化。而激活PI3K/AKT信號通路可減少Tau蛋白的Ser404和THr231位點磷酸化水平,進而改善AD。提示,激活PI3K/AKT信號通路可以改善Tau蛋白過度磷酸化。Tanabe等[17]的研究發現,在Tau蛋白過度磷酸化的AD模型鼠腦中,AKT活性下降且伴隨著GSK-3β活性增加。Eph-B2受體可通過激活PI3K/AKT信號通路,抑制GSK-3β活性,改善Tau蛋白過度磷酸化[18]。上述研究提示,PI3K/AKT信號通路可以通過抑制GSK-3β活性,改善Tau蛋白過度磷酸化。
研究發現急性有氧運動后短期內大鼠海馬組織中PI3K/AKT信號通路活性增加,GSK-3βSer9位點磷酸化水平升高,GSK-3β活性下降,其下游底物Tau蛋白的磷酸化水平也顯著降低;而48 h后,大鼠海馬組織中AKT和GSK-3β的磷酸化水平降低,Tau蛋白磷酸化恢復至基線水平[19]。提示急性運動后短期內激活PI3K/AKT信號通路可改善Tau蛋白的過度磷酸化,但具有時效性。長期運動訓練也可以激活PI3K/AKT信號通路,改善AD病理。3個月耐力運動上調了AD小鼠腦內PI3K和AKT的磷酸化表達,降低了AD小鼠海馬組織Tau蛋白磷酸化水平[20]。 房國梁等[21]發現,8周抗阻運動提高了大鼠大腦皮質和海馬組織PI3K的含量及活性,促進了AKT在Thr308和Ser473位點的磷酸化,活化的AKT通過磷酸化GSK-3βSer9位點,抑制GSK-3β活性,抑制了Tau蛋白多個位點的過度磷酸化。以上研究均說明,運動可以通過激活PI3K/AKT信號通路,促進GSK-3β磷酸化,抑制GSK-3β活性,緩解Tau蛋白過度磷酸化,改善AD認知功能障礙。
1.3 運動激活PI3K/AKT改善神經炎癥
已有研究證實,AD轉基因小鼠腦內存在胰島素抵抗,胰島素抵抗可導致機體巨噬細胞、平滑肌細胞等產生大量炎癥因子,如白介素-1(Interleukin-1,IL-1)、核轉錄因子κB(Nuclear FactorκB,NF-κB)和腫瘤壞死因子-α(Tumor Necrosis Factor-α,TNF-α)等,發生炎癥反應。毛小元等[22]研究發現,應用蛇床子素(OST)治療糖尿病腦病大鼠后,降低了大鼠海馬組織中炎癥因子NF-κB、TNF-α和IL-1β,加入PI3K抑制劑后,加重了炎癥反應,提示炎癥反應與PI3K/AKT信號通路有關。硝唑尼特(Nitazoxanide,NTZ)處理LPS誘導的BV2細胞后,激活了PI3K和AKT,增加了IκB表達,并抑制了NF-κB的核易位[23]。表明NTZ降低炎癥反應是通過PI3K/AKT/IκB/NF-κB途徑實現的。同時,NTZ治療APP/PS1小鼠后,小鼠腦中促炎因子IL-1β、TNF-α和誘導型一氧化氮合酶(Inducible Nitric Oxide Synthase,iNOS)表達顯著降低,提示NTZ可能通過PI3K/AKT信號通路降低小鼠腦中的促炎因子。
研究表明,游泳運動激活了D-半乳糖誘導AD大鼠海馬組織中IGFI-R/PI3K/AKT信號通路,且降低了炎癥因子TNF-α、p-NF-κB、環氧化酶(Cyclooxygenase,COX-2)和iNOS的蛋白水平,提示運動可能通過激活PI3K/AKT信號通路,降低海馬組織內的炎癥反應[24]。另有Wang等[25]研究發現,有氧運動激活了糖尿病大鼠前額葉中PI3K/AKT信號通路,增加了下游因子FOXO1的磷酸化,降低FOXO1乙酰化水平及活性,抑制其下游NF-κB表達,從而抑制炎癥反應。
1.4 運動激活PI3K/AKT增加自噬水平
PI3K/AKT信號通路下游的靶蛋白mTOR是調節自噬的主要因子,其主要復合物mTOR復合物1(mTOR complex1,mTORC1)參與調節細胞生長、凋亡、能量代謝和細胞自噬,活化的mTORC1可以抑制自噬并促進神經元中的蛋白質合成。不同刺激物(例如胰島素、IGF、生長因子和氨基酸等)可以激活PI3K-AKT信號通路,通過結節性硬化癥蛋白復合體(Tuberous Sclerosis Complex,TSC)抑制mTORC1上游信號蛋白Rheb,從而抑制mTORC1生成,進而誘導自噬過程。有研究表明mTOR誘導的自噬活動與AD 2種病理過程(Aβ沉積和Tau蛋白過度磷酸化)密切相關。研究發現,Aβ沉積加劇mTOR信號傳導,而抑制mTOR信號可降低Aβ水平[26]。此外,Shen等[27]發現,應用mTOR抑制劑抑制mTOR活性可改善Tau蛋白過度磷酸化。因此,激活PI3K/AKT信號通路可以抑制下游分子mTOR表達,調控AD腦內自噬,減少Aβ沉積和Tau蛋白過度磷酸化,改善AD認知。
先前研究指出,12周跑臺運動激活了自噬水平,減少了APP/PS1轉基因小鼠海馬組織中Aβ沉積[28]。同時,運動可以通過激活PI3K/AKT信號通路,緩解AD病理[29]。推測運動可通過激活PI3K/AKT通路激活自噬,促進Aβ清除。Kang等[30]研究發現,NSE/htau23轉基因AD小鼠大腦皮層中mTOR磷酸化異常,自噬標志性蛋白Beclin-1和LC3B減少,自噬受損,而12周跑臺運動激活了PI3K/AKT信號通路,抑制其下游分子mTOR活性,增加自噬蛋白Beclin-1表達,增強自噬活性,延緩AD小鼠認知功能下降。因此,運動可激活PI3K/AKT通路,抑制mTOR活性,增加AD腦內自噬水平,緩解AD癥狀。
綜上,運動可以激活PI3K/AKT信號通路調控下游分子GSK-3β、GSK-3α、NF-κB和mTOR等,從而降低Tau蛋白過度磷酸化、減少Aβ沉積、緩解炎癥反應和增強自噬水平,改善AD認知能力。
2 MAPK信號通路與AD
MAPK信號通路是哺乳動物細胞中的重要胰島素信號通路。MAPK信號通路可以被生長因子、細胞因子、激素和細胞應激等細胞外刺激激活,繼而觸發三級酶促級聯反應(MAPKKK-MAPKK-MAPK),參與細胞生長、分化和凋亡等生理過程[31]。目前在哺乳動物細胞中發現3條MAPK信號通路:細胞外信號調節蛋白激酶(Extracellular Signal-regulated Kinase,ERK)、c-Jun N末端激酶(c-Jun N-terminal Kinase,JNK)和p38絲裂原活化蛋白激酶(p38 Mitogen Activated Protein Kinases,p38MAPK),這3條通路與AD特征性病理改變有密切關系。
2.1 運動調控MAPK減少Aβ沉積
p38MAPK是MAPK家族中介導細胞存活和凋亡的重要通路。研究發現,AD小鼠腦內p38MAPK活化水平顯著升高[32]。敲除AD模型小鼠p38αMAPK的編碼基因mapk14,降低p38αMAPK表達,發現β-分泌酶1(β-site APP cleaving enzyme1,BACE1)溶酶體降解,BACE1表達降低,AD小鼠腦內Aβ生成減少。提示抑制p38MAPK表達,可降低BACE1表達和活性,抑制Aβ生成。除了p38MAPK信號通路,細胞外調節蛋白激酶 (Extracellular Regulated Protein Kinase,ERK)信號通路也是MAPK經典信號轉導通路。AD鼠海馬組織內,ERK磷酸化水平降低,Aβ沉積增加[33]。而增加APP/PS1小鼠海馬組織中ERK磷酸化水平,可以抑制APP和BACE1基因表達,減少Aβ沉積[34]。提示提高ERK信號通路活性可通過抑制APP和BACE1基因表達,減少Aβ生成。JNK是MAPK的一種亞類,又被稱為應激活化蛋白激酶(SAPK),近期有研究發現,c-Jun氨基末端激酶(c-Jun Nterminal Kinase,JNK)信號通路與AD發病密切相關,參與早期老年斑形成[35]。Savage等[36]用特異性活化抗體檢測不同基因鼠中的MAPK信號通路,證實激活JNK信號通路可導致Aβ沉積增加。喂養雄性TgCRND8轉基因小鼠4個月異丁香膽堿(IRN)抑制JNK信號通路后發現,Aβ沉積減少,小鼠認知功能得到改善[37]。 綜上,在AD中,抑制p38MAPK和JNK信號通路,激活ERK信號通路可減少Aβ沉積。
研究發現,跑臺運動抑制了AD模型鼠腦內MAPK通路(p38和JNK),提高了認知功能[38]。此外,急性跑臺運動上調了高脂膳食誘導肥胖小鼠腦內ERK的表達,降低了BACE1活性,減少了Aβ沉積,而JNK和p38MAPK表達未有顯著改變[39]。提示,急性跑臺運動可以通過激活ERK通路,減少Aβ沉積,但急性運動對JNK和p38MAPK通路無顯著作用,可能受長期運動影響才能實現對Aβ清除。而Pena等[40]發現,9周抗阻訓練降低了3xTg-AD小鼠海馬組織中APP水平,但未改變ERK蛋白含量及磷酸化水平。提示,運動激活ERK信號通路減少Aβ沉積,可能與運動類型有關。綜上,未來仍需進一步探究不同運動類型及運動周期干預MAPK(JNK、p38MAPK、ERK)信號通路從而減少Aβ沉積的作用機制。
2.2 運動調控MAPK改善Tau蛋白過度磷酸化
Tau蛋白Thr231位點過度磷酸化可激活MAPK信號通路,促進神經元中細胞周期激活機制,導致細胞死亡[41]。同時,JNK、p38和ERK等幾種激酶也可使Tau蛋白過度磷酸化。APP二聚化可激活ASK1-MKK6-p38級聯反應,使Tau過度磷酸化,導致認知功能障礙[42]。GSK-3是Tau蛋白磷酸化的重要激酶,有研究發現,下調B103-695細胞中APP表達可激活Ras-ERK信號通路,抑制GSK-3活性,降低Tau蛋白過度磷酸化[43]。提示激活ERK信號通路,可降低Tau蛋白過度磷酸化。另有研究發現,藥物利拉魯肽治療AD轉基因小鼠后,激活了ERK信號,抑制了JNK表達,改善了Tau蛋白高磷酸化水平[44]。以上研究說明,抑制p38MAPK、JNK通路和激活ERK通路,可改善Tau過度磷酸化,進而調控AD認知障礙。
運動可以改善AD腦內Tau蛋白過度磷酸化,其機制可能與海馬組織中的MAPK信號通路有關。Wang等[45]發現,12周跑臺運動降低了JNK和p38MAPK磷酸化水平,提高了ERK1/2磷酸化水平,減少了Tau蛋白過度磷酸化,改善了AD轉基因小鼠認知功能障礙。另外,12周跑臺運動增加了24月齡Tg-NSE/PS2m AD小鼠海馬組織內ERK磷酸化水平,降低了JNK和p38MAPK磷酸化水平,降低了Tau蛋白在Ser404、Ser202和Thr231位點的磷酸化水平[46]。提示運動通過抑制JNK、p38MAPK信號通路和激活ERK信號通路,降低Tau蛋白磷酸化,從而改善AD認知功能。
2.3 運動調控MAPK改善神經炎癥
AD中存在神經炎癥,且與MAPK信號通路有關。研究發現,Vitegnoside抑制了p38MAPK和JNK信號通路,減少了下游NF-κB炎癥信號轉導,緩解了AD細胞模型中的炎癥反應[47]。另有研究發現,中藥苓桂術甘湯治療AD大鼠顯著抑制了AD大腦中p38表達,上調了ERK信號,降低了NF-κB和IκBα表達,減少了促炎細胞因子產生[48]。以上研究說明,抑制p38MAPK和JNK信號通路,激活ERK信號通路,可降低下游炎癥信號NF-κB轉導,減少炎癥因子表達,緩解AD神經炎癥。
研究發現,跑臺運動下調了早老素2突變小鼠海馬組織中的ATF6α和sXBP1的表達,抑制了小鼠海馬組織中JNK和p38MAPK信號通路,降低了海馬組織中炎癥因子TNFα和IL-1α表達,改善了小鼠認知障礙[49]。另有實驗發現,運動抑制了AD模型鼠海馬組織中JNK和p38MAPK信號通路,激活了ERK信號通路,降低了海馬中TNF-α和IL-1β水平[38]。因此,運動可通過抑制海馬組織中的JNK和p38MAPK信號通路,激活ERK信號通路來降低炎癥反應。
2.4 運動調控MAPK改善自噬水平
脂多糖(Lipopolysaccharide,LPS)降低了小膠質細胞BV2中自噬標記物LC3-II的表達,并且LPS誘導了p38αMAPK激活,而SB203580(p38αMAPK抑制劑)處理后,發現LPS誘導的p38αMAPK活化被抑制,LC3-II表達顯著上升[50]。提示抑制p38MAPK信號通路會提高自噬水平。在AD小鼠腦內皮層和海馬神經元中特異缺失p38αMAPK,LC3-II和Beclin-1表達顯著升高,提高了AD小鼠腦內自噬水平[51]。綜上,可通過抑制p38MAPK通路,增加下游相關自噬因子LC3-II和Beclin-1表達,增加AD內自噬水平。
研究發現,遞增負荷訓練下調了衰老小鼠中轉化 生 長 因 子-β1(Transforming Growth Factor-β1,TGF-β1)及其下游信號分子TGF-β活化激酶1(TAK1)、MAPK、MKK3和p38MAPK的 表 達,且E-cadherin,Beclin-1和LC3的蛋白表達水平顯著增加[52],提示遞增負荷訓練可通過抑制TGF-β1/TAK1/MMK3/p38MAPK信號通路,激活自噬水平,從而改善老年小鼠的腎纖維化。因此,猜測運動可能會通過抑制p38MAPK信號通路提高AD自噬水平,改善AD認知障礙。目前尚無運動干預AD模型調控MAPK信號通路改善自噬的相關研究,未來該方向有待進一步研究。
3 Wnt信號通路與AD
Wnt信號通路在神經元分化、遷移、突觸發生等過程中起關鍵作用。其中,Wnt/β-連環蛋白信號通路(canonical Wnt/β-catenin)作 用 最 為 廣 泛,該 通 路中,Wnt蛋白是重要的啟動因子,當Wnt蛋白不存在時,Wnt/β-catenin信號通路被抑制,胞內的支架蛋白Axin、APC與GSK-3β和Ⅰ型酪蛋白激酶(Caseinkinase1,CK1)形成破壞復合物,介導β-catenin磷酸化,泛素介導的蛋白酶體識別并降解β-catenin,導致Wnt/β-catenin途徑中斷。 當Wnt蛋白存在時,Wnt/β-catenin信號通路被激活,Wnt與細胞表面受體(Frizzled)和膜輔助受體-低密度脂蛋白受體相關蛋白(LRP5/6)結合,使散亂蛋白與Axin結合,破壞Axin/APC/GSK-3β/CK1形成的復合物,抑制β-catenin的磷酸化[53]。β-catenin進入細胞核并與轉錄因子(Lymphoid Enhancer-Binding Factor 1,LEF1)/T細胞因子(T Cell Factor,TCF)結合,引起Wnt靶基因的轉錄,參與生物體的生長、發育以及分化等活動。研究發現,Wnt/β-catenin信號通路與多種神經系統疾病的發生和發展有關,尤其是與AD的發病密切相關。Wnt/β-catenin信號通路可調控Aβ的生成與清除、Tau蛋白過度磷酸化、神經炎癥以及自噬等影響認知功能。
3.1 Wnt與Aβ
在AD患者及動物模型中,Aβ的生成和清除與Wnt/β-catenin信號通路有關。CHO 7PA2細胞實驗發現,抑制Wnt/β-catenin信號通路,Aβ42濃度增加,Aβ42/Aβ40比率和低分子量Aβ低聚物(如三聚體和四聚體)增加,提示阻斷Wnt信號可導致APP發生淀粉樣蛋白水解,增加C99和Aβ42表達[54]。動物實驗發現,抑制J20TgAD小鼠Wnt/β-catenin信號通路,增加了小鼠海馬組織Aβ42產生,小鼠記憶力受損[55]。進一步研究發現,在Wnt/β-catenin信號通路中,Aβ可與GSK-3β結合,降解β-catenin引起神經細胞毒性,加重認知障礙[56]。同時,腦內沉積的Aβ又可通過上調Dickkopf1(DKK1)表達抑制Wnt/β-catenin信號通路,導致Aβ清除速率進一步降低,加重AD病情[57]。在APP/Tau/PS1小鼠中,通過氟西汀增加β-catenin含量、抑制GSK-3β活性,可以減少APP裂解及Aβ生成,起到神經保護作用[58]。由此可知,激活Wnt信號通路可抑制Aβ產生,改善AD認知功能障礙。
3.2 Wnt與Tau蛋白
在J20TgAD小鼠中,Wnt信號功能障礙會加速Tau蛋白位點Thr231,Ser235和AT8的磷酸化,且發現阻斷Wnt途徑的野生型小鼠中也存在Tau蛋白過度磷酸化[55]。有研究指出,Wnt/β-catenin信號通路可以通過調控GSK-3β活性,調節Tau蛋白過度磷酸化[59]。進一步研究發現,成年轉基因小鼠大腦中GSK-3β過度磷酸化導致β-catenin的核含量降低,增加了Tau蛋白磷酸化[60]。另有實驗發現,ANDRO激活Wnt信號可改善年輕和APP-PS1小鼠的Tau蛋白過度磷酸化[61]。進一步研究發現,ANDRO激活Wnt信號,降低了Tau蛋白位點Thr231、Ser235以及Ser202和Thr205(AT8表位)的磷酸化水平[62]。WASP-1可激活APP-PS1小鼠Wnt信號通路,抑制Tau蛋白PHF-1(Ser396和Ser404)位點磷酸化[63]。提示激活Wnt信號通路可通過抑制GSK-3β過度磷酸化,降低其活性,改善Tau蛋白過度磷酸化。
3.3 Wnt與神經炎癥
正常情況下,Wnt/β-catenin信號通路在炎癥誘導的免疫應答中發揮作用,其改善神經炎癥的機制是通過與其他通路的交叉作用來實現的。β-catenin是控制抗炎基因表達的激活物,過氧化物酶體增殖物激活受體γ(Peroxisome Proliferator-Activated Receptor Gamma,PPARγ)作為β-catenin的靶基因可以通過降低GSK-3β活性,激活Wnt信號通路,發揮炎癥調節作用[64]。GSK-3β同時參與調節NF-κB信號通路,參與神經炎癥發生。霍江濤等[65]發現川芎嗪激活AD大鼠腦內Wnt信號通路改善了大鼠腦內神經炎癥。此外,大麻二酚也是通過抑制GSK-3β活性,激活Wnt信號通路,緩解神經炎癥[66]。以上研究提示,抑制GSK-3β,可以激活Wnt信號通路,抑制NF-κB信號通路,從而改善神經炎癥。
3.4 Wnt與自噬
大量研究表明,激活Wnt可使機體內細胞自噬水平發生變化。Wnt3a配體促進了大鼠海馬組織CA1區自噬小泡的形成[67]。另有實驗發現,腦外傷小鼠鼻內注射Wnt3a后,提高了小鼠大腦內Wnt3a和β-catenin表達水平,下調了自噬蛋白Beclin-1和LC3-II表達,并改善了顱腦損傷小鼠運動能力[68]。腦外傷(TBI)損害機體的認知和記憶能力,藥物桑色素治療TBI大鼠模型后,增強了Wnt-1和自噬相關標記物(LC3B II/I和Beclin-1)蛋白表達,改善了小鼠學習記憶能力[69]。目前尚未有文獻直接說明AD中Wnt信號通路與自噬之間的關系,但根據以上實驗推測,在AD模型中,激活Wnt信號通路可能參與調節AD中自噬活動,使得機體自噬水平正常化。
綜上,激活Wnt信號通路,可以抑制Aβ產生、改善Tau蛋白過度磷酸化、減輕神經炎癥,并且可使機體自噬水平正常化,改善AD認知功能障礙。
3.5 運動激活Wnt通路改善AD癥狀
12周跑臺運動增加了STZ誘導認知障礙小鼠海馬組織中Wnt-3表達,降低了GSK-3β表達,改善了小鼠認知功能障礙[70]。8周跑臺運動提高了大鼠大腦皮質和海馬組織中Wnt蛋白含量,增加了β-catenin穩定性,提高了Wnt受體蛋白LRP5/6含量,降低了Axin1和CK1的mRNA和蛋白水平,抑制了GSK-3β活性,說明運動激活Wnt信號通路,減少了β-catenin降解,有利于β-catenin進入細胞核完成轉錄,從而提高大腦學習和記憶能力[71]。另有研究亦發現,自主跑輪運動同樣可以激活Wnt信號通路,增加海馬組織DG區神經營養因子(Brain-Derived Neurotrophic Factor,BDNF)以及胰島素樣生長因子-1(Insulin-Like Growth Factor-1,IGF-1)表達,促進了海馬組織DG區神經發生[72]。進一步研究發現,8周的認知訓練增加了Tg2576大鼠海馬組織中β-catenin水平,降低了Tau蛋白的過度磷酸化。以上研究表明,運動可通過激活Wnt信號通路,增加β-catenin水平,抑制GSK-3β,降低Tau蛋白過磷酸化,改善AD認知功能障礙。
4 小結與展望
綜上研究,3條胰島素信號通路與AD的發病機制密切相關。運動可通過胰島素信號通路改善AD病理:運動可激活PI3K/AKT信號通路,抑制下游GSK-3β、GSK-3α、NF-κB、mTOR等分子,減少Aβ沉積,改善Tau蛋白過度磷酸化和神經炎癥以及增加自噬水平,緩解AD病癥;運動通過抑制p38MAPK和JNK信號通路,激活ERK信號通路,可降低Aβ沉積等癥狀;運動還可激活Wnt信號通路,增加β-catenin水平,從而改善AD認知功能障礙(圖1)。
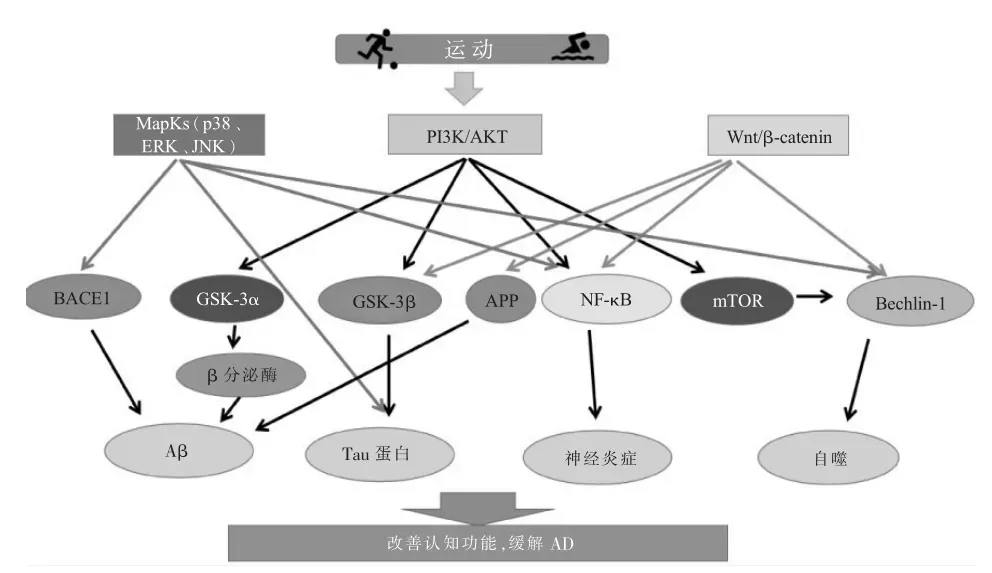
圖1 胰島素信號通路在運動改善阿爾茨海默病可能機制圖Figure1 Possible mechanism of insulin signaling pathway in the improvement of Alzheimer's disease by exercise
胰島素信號通路可能是運動改善AD的關鍵通路,但目前運動對AD中胰島素信號通路調節的研究還不夠全面,尤其是AD中MAPK及Wnt信號通路上下游分子的研究較少。此外,運動時間、運動強度、運動方式等因素對胰島素信號通路的影響都需進一步深入研究。未來可進一步明晰運動對AD中胰島素信號通路的調控作用和變化機制,深入探討改善AD的最佳運動模式,以期為運動防治AD提供更多理論數據。
猜你喜歡
作文周刊·小學二年級版(2022年20期)2022-05-05 01:33:06
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
學苑創造·A版(2020年9期)2020-10-13 09:41:02
創新作文(小學版)(2019年10期)2019-09-25 08:12:28
電子制作(2018年11期)2018-08-04 03:25:42
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
小學生學習指導(低年級)(2017年5期)2017-05-04 04:14:38
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
作文與考試·小學高年級版(2015年17期)2015-05-30 10:48:04
