Mn對中錳鋼α-Fe/γ-Fe界面影響的第一性原理研究
2021-09-25 11:53:58陳萬超張恒華
上海金屬 2021年5期
關鍵詞:界面
陳萬超 汪 楊 楊 晴 張恒華 張 梅
(上海大學材料科學與工程學院,上海 200444)
隨著我國人民生活水平的大幅度提高,私家車的保有量不斷增長,由此衍生的環(huán)境、能源和安全三大問題成為開發(fā)汽車用先進高強度鋼的主要推手。中錳TRIP鋼是一種具有高強塑性的高強鋼,其減重效果雖然不及鋁合金和鎂合金等,但是卓越的強塑性使它在汽車制造業(yè)始終占據一席之地[1-3]。
中錳TRIP鋼的原始組織為馬氏體,經過奧氏體逆相變退火并冷卻至室溫得到鐵素體和殘留奧氏體雙相組織。在逆相變退火過程中,部分馬氏體轉變成奧氏體,剩余的馬氏體由于碳元素的擴散使其碳含量不再飽和而轉變成鐵素體,隨后發(fā)生的錳元素配分起到穩(wěn)定奧氏體和細化鐵素體晶粒的作用[4];該過程中也有碳化物的形核析出,起沉淀強化作用,前人對其已有充分的研究。Jung等[5]采用第一性原理計算了FCC-Fe/MCs(NaCl結構,M=Ti,Zr,Hf,V,Nb,Ta)體系的共格界面能和半共格界面能,得出FCC-Fe/MCs體系的共格界面能低于BCC-Fe/MCs體系,BCC-Fe/MC體系的半共格和共格界面能隨失配量的增加而增大,奧氏體鋼中VC的沉淀強化效果最好。熊輝輝[6]采用第一性原理計算研究了鐵原子在(A1-xMx)C(A=Nb,Ti;M=Mo,V)復合碳化物以及A(C1-xNx)(A=Nb,Ti)復合碳氮化物的(001)表面的吸附行為,發(fā)現TiC(或NbC)晶格中的Ti(Nb)被Mn、V取代后形成的復合碳化物均有利于形核,Fe在(Nb0.5Mo0.5)C(001)表面具有最大吸附能和最短Fe—C鍵長。
Mn是中錳鋼的主要合金元素[4],錳的加入會促進逆相變退火過程中奧氏體的形核,導致室溫下殘留奧氏體量增加。在奧氏體逆相變過程中,奧氏體在馬氏體板條間長大,同時碳和錳在兩相間配分[7]。為了研究該過程中Mn配分對兩相區(qū)奧氏體的體積分數和穩(wěn)定性的影響,本文建立了鐵素體/奧氏體(α-Fe/γ-Fe)兩相界面,采用第一性原理計算方法分析了α-Fe/γ-Fe界面穩(wěn)定性和界面電子結構。
1 計算方法與模型建立
1.1 計算方法
本文計算均采用基于密度泛函理論[8](density functional theory,DFT)結合平面波贗勢方法的CASTEP(Cambridge sequential total energy package)軟件包,使用超軟贗勢描述原子核與電子之間的相互作用。為了得到合適的截斷能(Ecut)和k點,對α-Fe(體心立方,空間群IM-3M)與γ-Fe(面心立方,FM-3M)的結構進行了單點能收斂測試;為了得到合適的電子交換-相關泛函,選取3種電子交換關聯泛函(GGA-PBE,GGA-PW91,LDA-CAPZ)對α-Fe和γ-Fe的結構進行優(yōu)化計算,其晶胞參數的計算結果如表1所示。
對比表1的計算結果,本文計算采用的電子交換-相關泛函為GGA-PBE,所有計算在倒易空間上進行,第一布里淵區(qū)積分采用Monkhorst-Pack方案形成的特殊k點方法。對于體相α-Fe與γ-Fe,k點網格劃分為9×9×9;對于所有表面和界面模型,k點劃分均為9×9×1。最大平面波截斷能為400 eV,采用Broyden-Fletcher-Goldfarb-Shanno(BFGS)算法進行幾何優(yōu)化,實現原子的充分弛豫。收斂條件是自洽計算最終兩個循環(huán)能量之差小于1×10-5eV/atom,作用在每個原子上的力不大于0.03 eV/?,內應力不大于0.05 GPa。
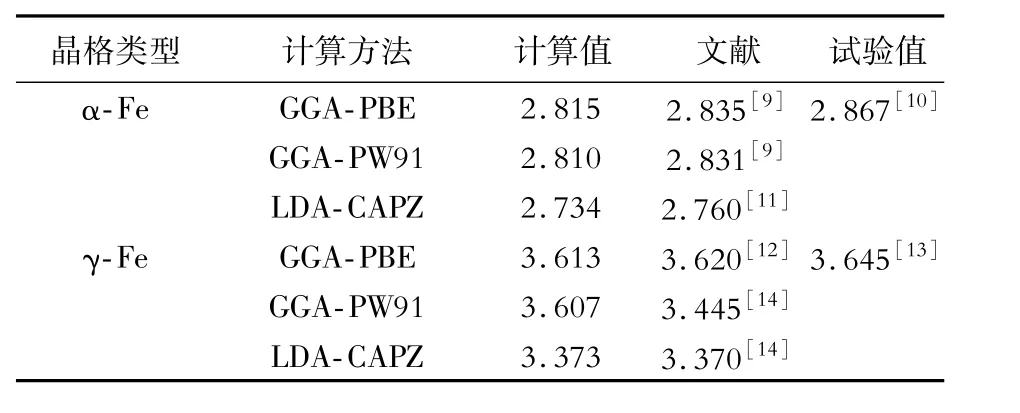
表1 α-Fe與γ-Fe點陣常數的計算值與試驗值Table 1 Calculated and experimental values of lattice constants of α-Fe and γ-Fe?
1.2 表面模型優(yōu)化
鐵素體與奧氏體滿足K-S取向關系:α-Fe(110)/γ-Fe(111)[15]。為了確定奧氏體和鐵素體表面的最小原子層數,保證構建界面的兩側Fe原子表面深處呈現體相原子的特征,對α-Fe(110)和γ-Fe(111)表面模型進行收斂性測試。這兩個表面模型均為非極性的特征,因此可根據表面能隨原子層數的變化來確定所需原子層數,其表面能[16]可根據式(1)計算得出:
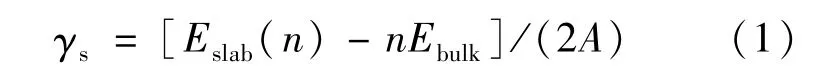
式中:Eslab(n)和Ebulk分別表示該相表面結構的總能量和組成表面結構單個原子(或分子)的能量;n表示原子(或分子)的個數。表面能的計算結果如表2所示。
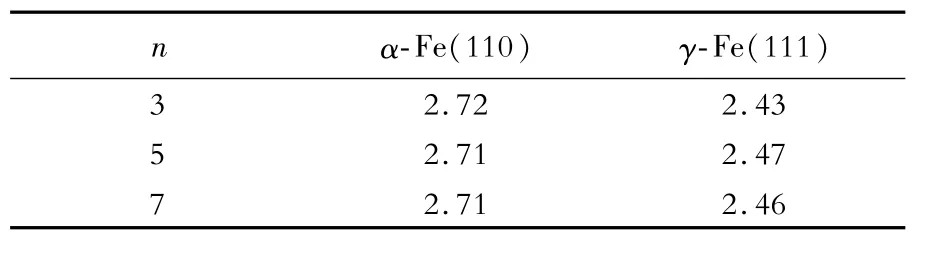
表2 α-Fe(110)和γ-Fe(111)的表面能收斂趨勢Table 2 Convergence trend of surface energy of α-Fe(110)and γ-Fe(111)J/m2
從表2可以看出,當n≥5時,α-Fe(110)和γ-Fe(111)表面能(γs)分別收斂于2.71和2.46 J/m2。為了計算方便,選擇5層原子層厚度的α-Fe和γ-Fe構建界面。
1.3 α-Fe/γ-Fe界面模型優(yōu)化
根據α-Fe/γ-Fe的取向關系建立的界面模型如圖1所示,共20個Fe(10個α-Fe原子,10個γ-Fe原子),將5層γ-Fe(111)堆垛在5層α-Fe(110)表面上,并在上下表面分別添加15 ?的真空層以消除兩者之間的相互作用。經過晶胞優(yōu)化后,用Mn原子分別置換界面次近鄰層的α-Fe原子(Case 1)、最近鄰層的α-Fe原子(Case 2)和γ-Fe原子(Case 3)以及次近鄰層的γ-Fe原子(Case 4),此時界面系統(tǒng)Mn的質量分數約為5%;未進行Mn原子置換的界面記為Clean。
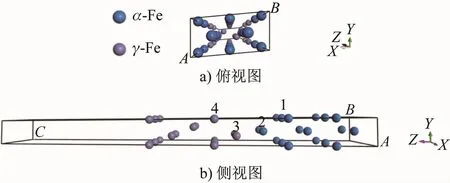
圖1 未馳豫的α-Fe/γ-Fe界面模型Fig.1 Model of the unrelaxed interface of α-Fe/γ-Fe
分離功[8](Wsep)在界面系統(tǒng)中定義為分離兩凝聚相形成各自表面所作的可逆功,計算公式為:
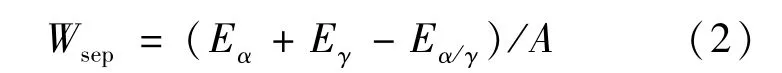
式中:Eα、Eγ分別表示弛豫的游離α-Fe和γ-Fe表面結構的總能量;Eα/γ表示該界面的總能量;A表示界面面積。在熱力學上,分離功越大,界面越穩(wěn)定。
兩固相的界面能[16](γint)計算公式為:
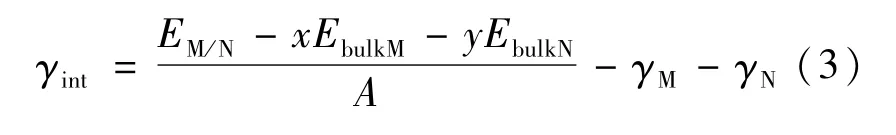
式中:EbulkM和EbulkN分別表示M(α-Fe)相和N(γ-Fe)相中單個原子(或分子)的能量,x和y是界面模型中兩相原子(或分子)的個數;γM與γN表示兩相的表面能;A表示界面面積。固相間的界面能定義為形成單位面積的界面時所增加的吉布斯自由能,但其本質上源于界面處的原子晶格畸變、化學鍵的改變和結構應變[7]。如果兩個不同的固相之間能夠形成一個穩(wěn)定的界面,則該界面的界面能應為正值,且界面能越小,在熱力學上越穩(wěn)定。
在經典形核理論中,晶界形核所需的臨界形核功G*和臨界形核半徑r*分別如式(4)和式(5)所示,可以看出,界面能越小,臨界形核半徑和臨界形核功越小,越有利于形核[14]。
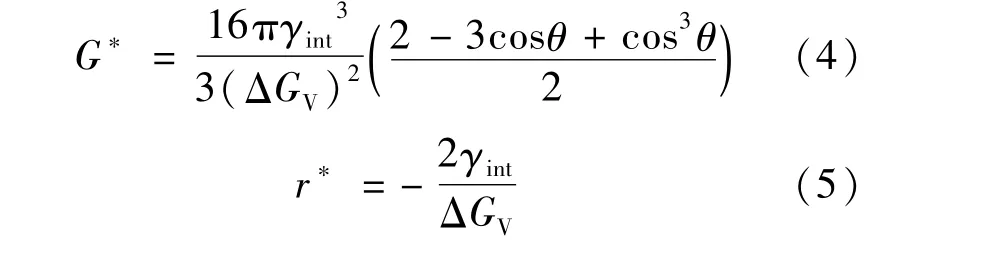
式中:γint為鐵素體與奧氏體之間的界面能;θ為晶核γ-Fe和α-Fe的接觸角;GV為體系的體積自由能。
2 計算結果與討論
2.1 Mn對界面穩(wěn)定性的影響
Mn原子置換前后界面系統(tǒng)的界面能和分離功如圖2和表3所示。可以看出,Mn原子的置換對界面性質有一定影響。當Mn的質量分數上升至5%時,界面系統(tǒng)的分離功(Wsep)從負值變?yōu)檎担–ase 1除外),界面穩(wěn)定性提高,奧氏體形核位點增加,其中置換界面次近鄰γ-Fe原子(Case 4)的分離功最大。置換界面最近鄰層的α-Fe原子(Case 2)、γ-Fe原子(Case 3)和次近鄰的γ-Fe原子(Case 4)的界面能(γint)相比Clean界面能均下降,界面更穩(wěn)定,與分離功變化趨勢一致。
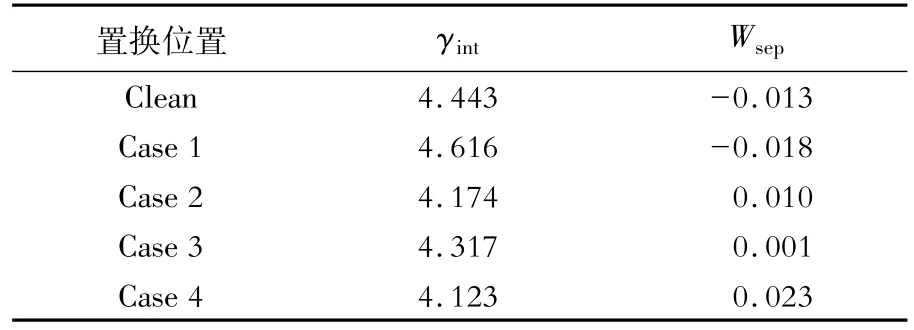
表3 Mn原子置換Fe原子前后界面系統(tǒng)的分離功和界面能Table 3 Separation work and interface energy of interface system before and after replacing Fe atom by Mn atomJ/m2
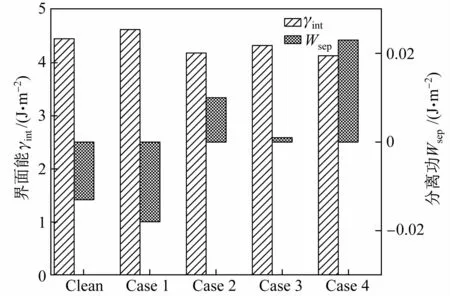
圖2 Mn原子置換Fe原子前后的界面系統(tǒng)的分離功和界面能變化Fig.2 Variation of separation work and interface energy of interface system before and after replacing Fe atom by Mn atom
界面能源于晶格畸變和化學鍵,而晶格畸變直接導致界面兩端鐵素體與奧氏體的晶格錯配度μ的變化,晶格錯配度的計算公式[17]如式(6)所示,根據式(6)計算的錯配度如表4所示。
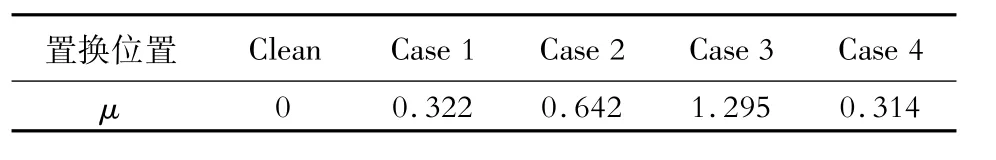
表4 Mn原子置換Fe原子后界面系統(tǒng)的晶格錯配度Table 4 Lattice mismatch degree of interface system after replacing Fe atom by Mn atom%
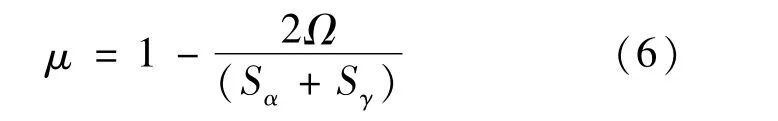
式中:Ω為α-Fe相表面和γ-Fe相表面封端重疊部分的面積;Sα和Sγ分別為界面兩端α-Fe和γ-Fe相表面封端的面積。
結合晶格錯配度,從熱力學角度解釋界面能的變化原因:α-Fe和γ-Fe形成界面兩相引起晶格畸變,導致晶格錯配度發(fā)生變化。但本文討論的是Mn原子進入界面產生的晶格畸變,因此把Clean的晶格錯配度定義為零;Case 1相比Clean界面能上升的原因可能是Mn原子進入奧氏體使得α-Fe和γ-Fe晶格錯配度從0提高到0.322%;Case 2、Case 3及Case 4的晶格錯配度分別為0.642%、1.295%、0.314%,但相比Clean界面能都下降,這可能是化學鍵變化所致。
從經典形核理論分析界面能的變化,Case 2、Case 3、Case 4的界面能相比Clean下降,說明這3種情況下奧氏體的形核功和形核半徑減小,奧氏體形核數量增加且形核尺寸更小。其中Case 4的奧氏體形核數量最多且最細小,奧氏體熱穩(wěn)定性最好。
2.2 Mn原子對界面鍵合的影響
Milliken布居分析可以半定量地描述電荷轉移量,用來判斷鍵能的變化[6]。圖3為Mn原子引入前后界面系統(tǒng)中各個原子的轉移電荷數量。可以看出,Mn原子無論置換哪個位置的Fe原子,置換后的Mn原子相較于原來的Fe原子轉移電荷數大量增加,Case 1~4中Mn的轉移電荷數分別為0.20、0.18、0.18和0.17,所以Mn原子會形成一個強烈的正電場,吸引附近其他Fe原子周圍的電子,形成共價鍵。在Clean中,界面最近鄰層的5號和15號α-Fe原子(界面最近鄰層的α-Fe原子)以及6號和16號γ-Fe原子(界面最近鄰層的γ-Fe原子)轉移電荷量都為0,表示此時界面兩側Fe原子相互作用較弱,界面鍵合較弱,保持亞穩(wěn)狀態(tài)(與負的分離功對應);Case 1中,當Mn原子置換14號α-Fe原子后,5號和15號α-Fe原子轉移電荷數變?yōu)椋?.04、-0.02,而6號和16號γ-Fe原子轉移電荷量仍為0,因此前者周圍的負電荷被Mn原子吸引,遠離了6號和16號γ-Fe原子,使得亞穩(wěn)態(tài)的界面變得更不穩(wěn)定,此時共價鍵作用與晶格畸變作用一致,使界面趨于不穩(wěn)定,因此在共價鍵與晶格畸變雙重作用下,界面能達到最高值;Case 2中Mn原子取代15號α-Fe原子后,6號和16號γ-Fe原子的轉移電荷數變?yōu)椋?.01和-0.06,被Mn原子產生的正電場所吸引,界面趨于穩(wěn)定,共價鍵作用與晶格畸變作用相悖且占據主導地位,即使晶格畸變相比Case 1進一步加劇,但界面能仍然下降;Case 3中Mn原子取代16號γ-Fe原子后,5號和15號γ-Fe原子的轉移電荷數變?yōu)椋?.02和-0.03,并被Mn原子吸引形成共價鍵使界面結合更穩(wěn)定,但此時共價鍵的作用比晶格畸變作用弱(Case 3相比Case 2的晶格畸變程度幾乎翻倍,如表4所示),晶格畸變占據主導地位,導致界面能上升;Case 4中Mn原子置換17號γ-Fe原子,5號α-Fe原子轉移電荷數變?yōu)椋?.01,6號和16號γ-Fe原子的轉移電荷數變?yōu)椋?.01和-0.04,此時界面兩側Fe原子周圍負電荷相互排斥使得界面變得不穩(wěn)定,但Case 4相比Case 3的晶格畸變程度大幅度減小,界面能又下降。
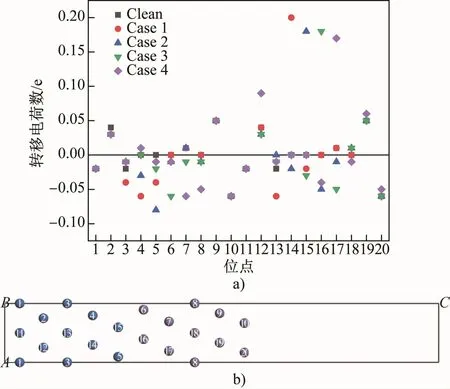
圖3 錳的質量分數為5%的界面系統(tǒng)中各個位點原子的轉移電荷數散點圖(a)及位點編號(b)Fig.3 Scatter diagram of the number of transfer charges of atoms at each site(a)and site number(b)in the interface system with 5% Mn by mass
為了進一步闡述界面的電子性質,計算了Mn原子引入前后界面系統(tǒng)的總態(tài)密度(TDOS,total density of states),如圖4所示。可見5種界面的總態(tài)密度曲線變化趨勢相似,說明Mn原子置換對界面能的影響不大。費米能級處的態(tài)密度可以反映界面的穩(wěn)定性[17],態(tài)密度越小說明界面越穩(wěn)定,且態(tài)密度不為零證明界面存在金屬鍵。從圖4可知,費米能級處的態(tài)密度大小順序為Case 4<Case 2<Case 3<Clean<Case 1,與界面能結果一致。此外,α-Fe(110)/γ-Fe(111)界面最顯著的特征是費米能級兩側的峰1和峰2之間的態(tài)密度不為零,這是一個贗隙[18]。這個寬的贗隙(-1.5~2 eV)表明在α-Fe(110)/γ-Fe(111)界面還存在強定向共價鍵,因此這個界面同時存在金屬鍵和共價鍵。
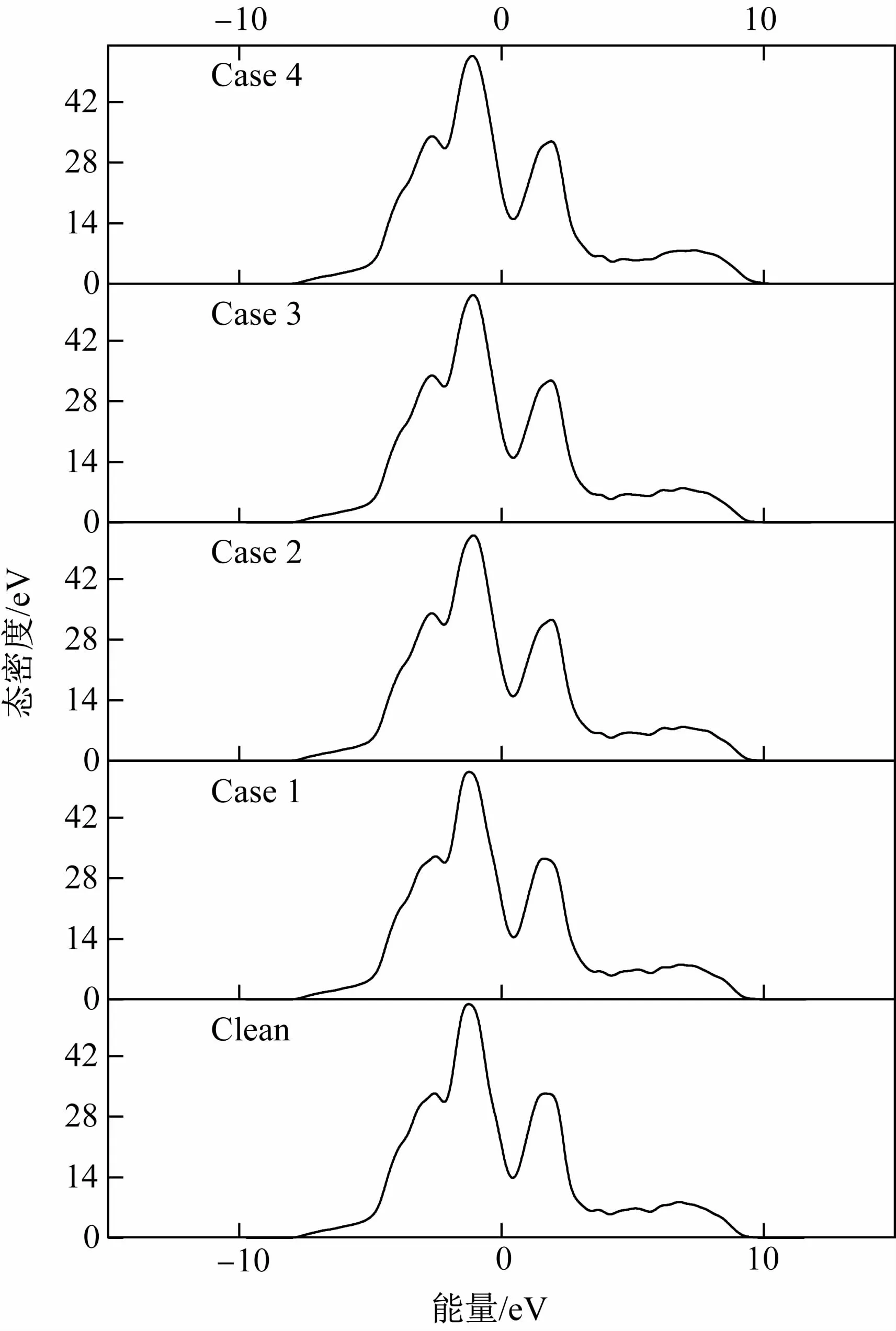
圖4 5種界面系統(tǒng)的總態(tài)密度Fig.4 Total density of states of five interface systems
3 結論
(1)γ-Fe(111)表面和α-Fe(110)表面均于5層達到收斂,表現出明顯的體相特征。
(2)Mn原子置換Fe原子后,一方面使界面兩側鐵素體和奧氏體之間的錯配度發(fā)生不同程度的變化,即界面的晶格畸變程度發(fā)生變化;另一方面使各個原子的轉移電荷數發(fā)生變化,即界面共價鍵和金屬鍵強度發(fā)生變化。晶格畸變程度和化學鍵強度變化共同作用使界面能發(fā)生變化。
(3)Mn的質量分數為5%時,Mn向奧氏體一側擴散使得α-Fe/γ-Fe界面更加穩(wěn)定,同時由于界面能的下降,奧氏體的臨界形核功和形核尺寸減小,奧氏體的形核數量和熱穩(wěn)定性提高。
猜你喜歡
艦船科學技術(2022年16期)2022-09-22 02:15:00
北京航空航天大學學報(2021年6期)2021-07-20 07:23:54
當代陜西(2020年13期)2020-08-24 08:22:02
制造技術與機床(2017年5期)2018-01-19 02:49:17
制造技術與機床(2017年11期)2017-12-18 06:47:29
金秋(2017年4期)2017-06-07 08:22:16
蘇州科技大學學報(自然科學版)(2017年1期)2017-03-20 15:25:18
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
