2-碘-3-(對甲苯磺酰氧基)苯基醚的選擇性合成
2021-09-17 10:09:30潘峰王磊郭怡沈金瀅潘曉峰鄭衛新
浙江大學學報(理學版) 2021年5期
潘峰,王磊,郭怡,沈金瀅,潘曉峰,鄭衛新
2-碘-3-(對甲苯磺酰氧基)苯基醚的選擇性合成
潘峰,王磊,郭怡,沈金瀅,潘曉峰,鄭衛新*
(杭州師范大學材料與化學化工學院,浙江 杭州 311121)
磺酰氧基鹵代芳基醚是一種多功能化合物,在眾多研究領域具有廣泛應用價值。以2-碘間苯二酚為原料,經化學計量的雙磺酰化、選擇性單側水解及與溴化物的親核取代反應,合成系列2-碘-3-(對甲苯磺酰氧基)苯基醚。結果表明,雙磺酰化產物的選擇性單側水解比2-碘間苯二酚通過單烴基化進行原料中2個對稱羥基的選擇性官能化更有優勢。以低成本堿為水解試劑,探索反應時間、溫度等因素對2,6-雙(對甲苯基磺酰氧基)碘苯單側選擇性水解的影響,水解產物核磁純度>99%,為對甲苯磺酰氧基碘代芳基醚的合成提供一種操作簡便、反應條件溫和、成本低和選擇性高的方法。所有化合物結構均經1H-NMR、13C-NMR與高分辨質譜等方法確定。
磺酰氧基碘代苯基醚;合成;選擇性水解
芳基醚是廣泛存在于許多天然產物[1-2]和藥物[3-5]分子中的重要骨架。芳環上官能團的選擇性化學轉化是芳基醚官能化的重要策略,因此多官能化芳基醚的選擇性構建在合成化學中具有重要意義。磺酰氧基碘代芳基醚是組建眾多具有生理活性分子的重要前體[6-10],其結構式為
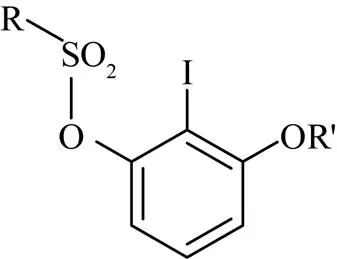
其中,相鄰的鹵素與磺酰氧基在芳基衍生化中發揮了重要作用[11-12]。鹵素與磺酰氧基的活性差異可為選擇性官能化提供有效途徑[13-17]。此外,該化合物是苯炔這一活性物種的重要供體,通過環加成反應參與多種活性物質的合成[6,18]。
磺酰氧基碘代芳基醚中因取代基性質差異而擁有2種活性不同的C—O鍵,對應苯二酚前體的選擇性反應是該類化合物合成的主要策略,實現酚羥基高選擇性烴化與磺酰化是合成該化合物的關鍵。其合成方法主要有2種,一是先通過鹵代苯二酚的非完全烴化反應進行單側烴基化,再進行磺酰化[19-22]合成;二是通過鹵代芳基二酚的磺酰化-選擇性水解-衍生化合成。其中,三氟磺酰氧基的反應較常見,例如1,6-雙(三氟磺酰氧基)碘苯可經Cs2CO3作用脫去其中一個磺酰基[23]。在常用磺酰基中,對甲苯磺酰基成本較低,近年來在許多過渡金屬催化的偶聯反應中表現出較好的活性和選擇性[24]。2,6-二羥基苯乙酮中的2個酚羥基可以通過控制計量的對甲苯磺酰氯進行選擇性單磺酰化,繼而在未反應的羥基鄰位發生碘代反應生成碘代丁二酰亞胺(NIS)[25]。CLARK等[26]報道了3,5-雙磺酰氧基碘苯的單側選擇性水解。然而,對于磺酰氧基碘代芳基醚的合成前體,即2-鹵代雙對甲苯磺酰二苯酯的磺酰化或選擇性水解,至今未見文獻報道。
基于此,本文采用低成本堿高選擇性地實現2,6-雙對甲苯磺酰氧基2-碘苯的單側水解,經親核取代反應生成磺酰氧基碘代芳基醚,為磺酰氧基鹵代芳基醚的合成提供一種低成本、高效的方法。
1 實驗
1.1 實驗儀器與實驗原料
Bruker Avance DMX500核磁共振儀,Bruker DMX300核磁共振儀,Shimadzu HRMS-EI-TOF型高分辨儀,顯微熔點儀X5。2-碘-1,3-苯二酚通過文獻[27]中方法合成。
1.2 2,6-雙磺酰氧基碘苯的合成
在250 mL燒瓶中加入2-碘代-1,3-苯二酚(4.72 g,20 mmol)、對甲苯磺酰氯(8.77 g,46 mmol)、碳酸鉀(11.06 g,80 mmol)和丙酮(100 mL),室溫下攪拌,薄層色譜(TLC)跟蹤至反應完全。抽濾,濾渣用二氯甲烷洗滌,合并的有機相用飽和食鹽水洗滌,經無水硫酸鎂干燥,旋蒸濃縮。用乙酸乙酯和石油醚對粗產物重結晶,得到白色晶體2,6-雙(對甲苯基)磺酰氧基碘苯。
1.3 2-碘-3-(對甲苯磺酰氧基)苯酚的合成
在1 L錐形瓶中加入2,6-雙磺酰氧基碘苯(23.3 g,42.8 mmol)和甲醇(100 mL),保持35 °C,滴加含KOH(5.0 g,89.1 mmol)的水-甲醇混合溶液(2.5 mL水,225 mL甲醇)。滴加完畢后保持溫度不變,攪拌3 h,升溫至45°C繼續攪拌15 min后冷卻至室溫,用水稀釋至總體積為800 mL,用濃度為5%的鹽酸中和,在4 °C下靜置2 d。過濾,將濾渣溶解在乙醚中,用10%的NaOH溶液萃取。分出的油狀物經乙醚洗滌后用5%的鹽酸中和。混合物用乙醚萃取3次,合并的有機相經硫酸鎂干燥,過濾,濃縮后得到2-碘-3-(對甲苯磺酰氧基)苯酚。
1.4 2-碘-3-(對甲苯磺酰氧基)苯基醚的合成
在250 mL燒瓶中加入2-碘-3-(對甲苯磺酰氧基)苯酚(7.80 g,20 mmol),鹵代烴(20 mmol),碳酸鉀(5.56 g,40 mmol)和乙腈(30 mL),室溫下攪拌,TLC跟蹤至反應完全。加50 mL水稀釋,用乙酸乙酯萃取混合物,合并的有機相用飽和食鹽水洗滌,無水硫酸鎂干燥,過濾。用快速柱層析分離(石油醚()∶乙酸乙酯()=10∶1)粗產物,提純得到系列2-碘-3-(對甲苯磺酰氧基)苯基醚。
2 結果與討論
2.1 2-碘間苯二酚的選擇性烴化
2-碘間苯二酚與芐溴的化學反應方程式為
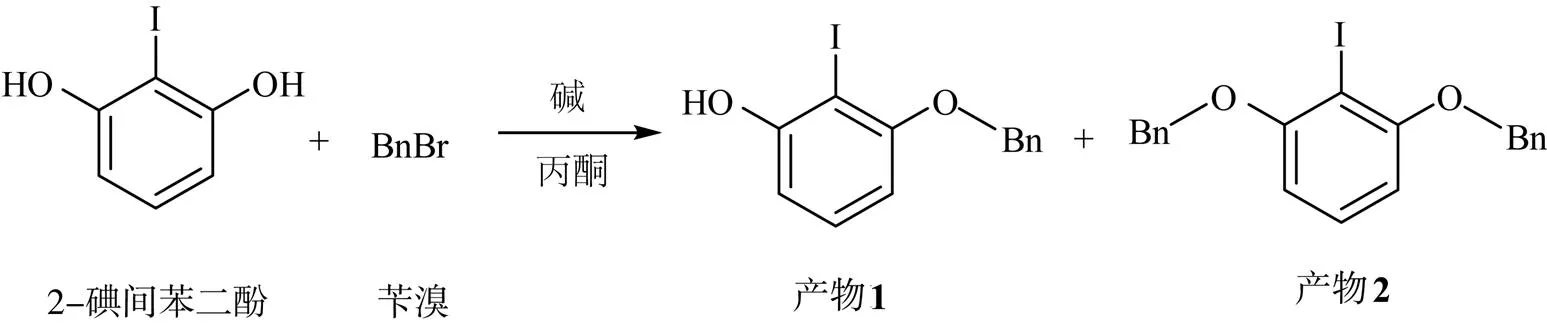
首先,選擇烴化反應作為單側選擇性取代的起點,室溫下將2-碘間苯二酚與堿在丙酮中混合,邊攪拌邊加入等物質的量的芐溴。不同堿的反應選擇性如表1所示。

表1 2-碘間苯二酚與芐溴的反應
a:堿與2-碘間苯二酚的物質的量之比為1.2∶1;b:以TLC跟蹤2-碘間苯二酚消失為準;c:核磁產率,括號內產物比例由反應混合物的1H-NMR確定,CH2Br2為內標。
由表1可知,在有機堿(Entry 4和5)作用下單取代產物1的產率高于無機堿(Entry 1~3)作用下產物1的產率,且可通過色譜柱分離產物1和2,但反應選擇性無法滿足高效合成的需求。嘗試用2-碘間苯二酚與等物質的量的對甲苯磺酰氯進行選擇性單側磺酰化,依然未獲得成功。
2.2 2, 6-雙磺酰氧基碘苯的選擇性水解
碘代間苯二酚在K2CO3作用下與對甲磺酰氯以物質的量之比為2∶1發生反應,計量生成2,6-雙磺酰氧基碘苯。該化合物在溶劑為甲醇,堿為NaOH[26]的條件下可實現單側水解,生成2-碘-3-(對甲苯磺酰氧基)苯酚,水解反應方程式為
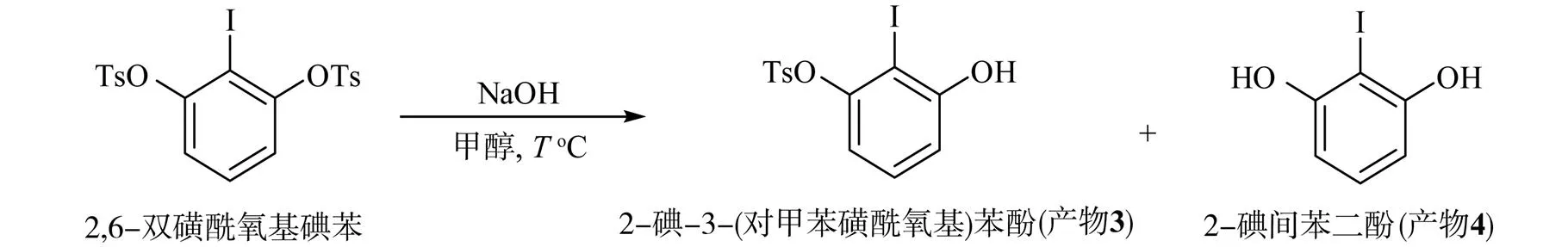
探索不同溫度下2,6-雙磺酰氧基碘苯的選擇性單側水解,結果見表2。
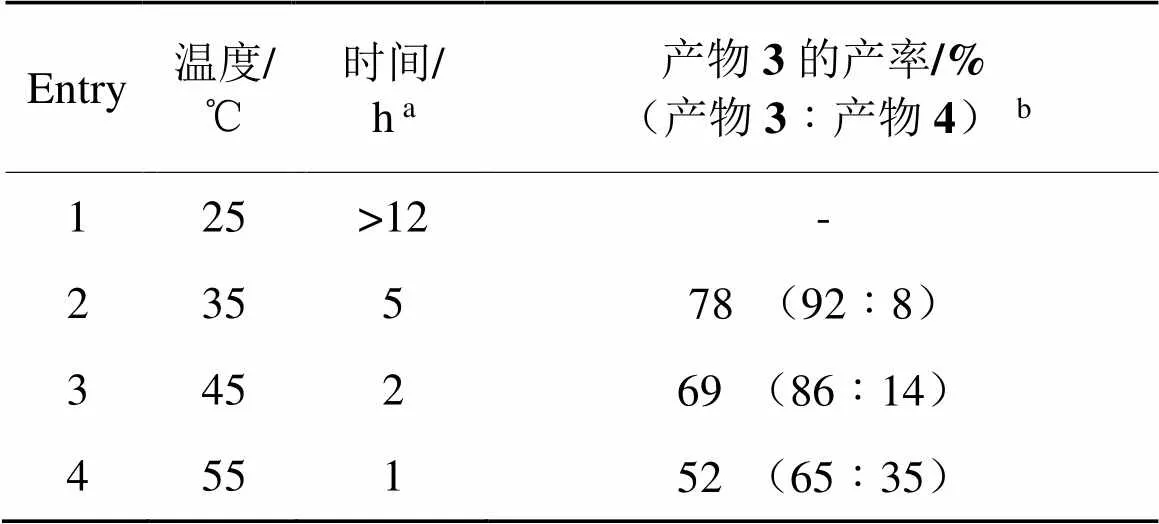
表2 2,6-雙磺酰氧基碘苯的選擇性單側水解
a:由于2,6-雙磺酰氧基碘苯不溶于反應溶液,此處時間是指從堿溶液滴完至固體消失的時間。b:核磁產率,括號內產物比例由反應混合物的1H-NMR確定,CH2Br2為內標。
由表2可知,在室溫25 ℃下,反應時間較長,即使2,6-雙磺酰氧基碘苯經歷12 h,仍未反應完全。溫度每升高10 ℃,反應明顯加快,完全水解產物增加。因此,先將溫度調至35 ℃反應3 h,然后升溫至45 ℃反應15 min,經后處理無須提純可獲得核磁純度>99%的2-碘-3-(對甲苯磺酰氧基)苯酚(產率達86%),如圖1所示。該產物可達到后續合成應用的需求。
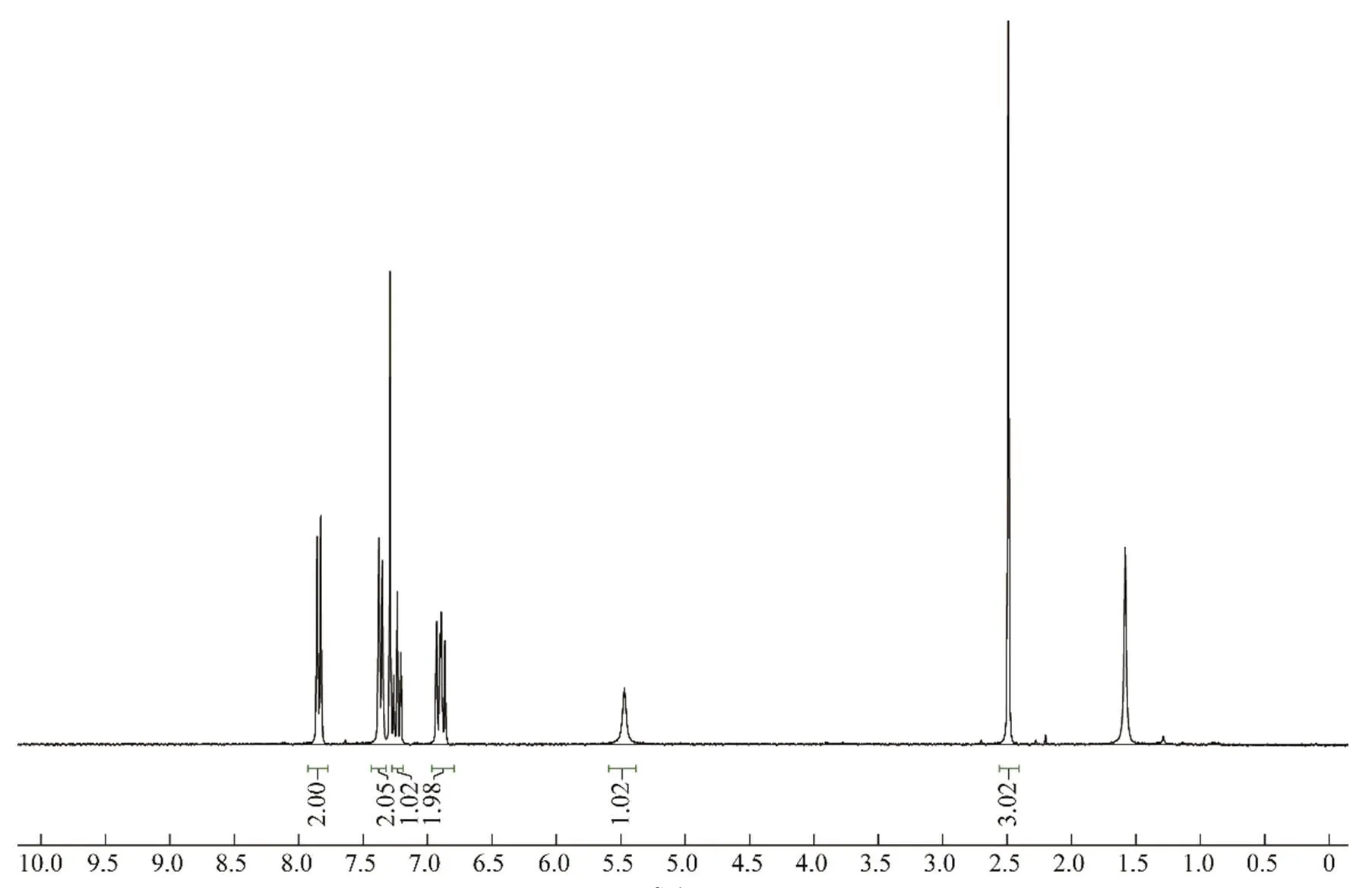
(ppm)

2.32-碘-3-(對甲苯磺酰氧基)苯酚的親核取代反應
以乙腈為溶劑,2-碘-3-(對甲苯磺酰氧基)苯酚在K2CO3作用下與多種溴代烴發生親核取代反應,生成系列2-碘-3-(對甲苯磺酰氧基)苯基醚(產物5a~j),其中,產物5a為甲基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為86%;產物5b為乙基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為78%;產物5c為正丙基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為79%;產物5d為異丙基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為88%;產物5e為芐基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為89%;產物5f為(2-苯基)乙基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為88%;產物5g為(2’-溴)芐基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為75%;產物5h為(2’-碘)芐基(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為81%;產物5i為烯丙基-(3-對甲苯磺酰氧基-2-碘)苯基醚,產率為82%;產物5j為(3-對甲苯磺酰氧基-2-碘)苯氧基乙酸乙酯,產率為83%。產物5a~j的詳細數據請掃描右側二維碼。
親核取代反應方程式為

產物5a~j的結構式如下:
3 結論
以2-碘間苯二酚為原料,經磺酰化、選擇性水解、親核取代反應,合成了系列磺酰氧基鹵代芳基醚。以低成本堿為水解試劑,實現2,6-雙(對甲苯基)磺酰氧基鹵代苯的單側選擇性水解,水解產物核磁純度>99%,為對甲苯磺酰氧基碘代芳基醚的合成提供了一種低成本、高效的方法。
[1]BRAD C, JOHN D F. Polybrominated diphenyl ethers from,and[J]. Tetrahedron, 1981, 37(13): 2335-2339. DOI:10.1016/s0040-4020(01)88886-4
[2]YAMADA T, TAKIGUCHI H, OHMORI K, et al. Total syntheses of pusilatins A-C, liverwort-derived macrocyclic bisbibenzyl dimers[J]. Organic Letters, 2018, 20(12): 3579-3582. DOI:10.1021/acs.orglett. 8b01366
[3]FUJIMOTO J, OKAMOTO R, NOGUCHI N, et al. Discovery of 3,5-diphenyl-4-methyl-1,3-oxazolidin-2-ones as novel, potent, and orally available delta-5 desaturase (D5D) inhibitors[J]. Journal Medicinal Chemistry, 2017, 60(21): 8963-8981. DOI:10.1021/acs.jmedchem.7b01210
[4]SINGER J M, WILSON M W, JOHNSON P D, et al. Synthesis and SAR of tolylamine 5-HT6 antagonists[J]. Bioorganic & Medicinal Chemistry Letters, 2009, 19(9): 2409-2412. DOI: 10.1016/j.bmcl.2009.03.077
[5]NORTON R S, CROFT K D, WELLS R J. Polybrominated oxydiphenol derivatives from the sponge dysidea herbacea[J]. Tetrahedron, 1981, 37(13): 2341-2349. DOI:10.1016/s0040-4020(01)88887-6
[6]MAMIKO N, YOSHIO A, FUMITAKA K, et al. Total synthesis of actinorhodin[J]. Angewandte Chemie International Edition, 2019, 58(13): 4264-4270. DOI:10.1002/anie.201814172
[7]BORGEL J, TANWAR L, BERGER F, et al. Late-stage aromatic C-H oxygenation[J]. Journal of the American Chemical Society, 2018, 140(47): 16026-16031.
[8]ARIYASU S, SAWA A, MORITA A, et al. Design and synthesis of 8-hydroxyquinoline-based radioprotective agents[J]. Bioorganic & Medicinal Chemistry, 2014, 22(15): 3891-905. DOI:10. 1016/j.bmc.2014.06.017
[9]BORGEL J, TANWAR L, BERGER F, et al. Late-stage aromatic C-H oxygenation[J]. Journal of the American Chemical Society, 2018, 140(47): 16026-16031.
[10]NING Y, FUKUDA T, IKEDA H, et al. Revisiting secondary interactions in neighboring group participation, exemplified by reactivity changes of iminylium intermediates[J]. Organic Biomolecular Chemistry, 2017, 15(6): 1381-1392. DOI:10.1039/c6ob02719a
[11]LIU Z, LI J, LI S, et al. SuFEx click chemistry enabled late-stage drug functionalization[J]. Journal of the American Chemical Society, 2018, 140(8): 2919-2925. DOI:10.1021/jacs.7b12788
[12]IQBAL J, EL-GAMAL M I, EJAZ S A, et al, Tricyclic coumarin sulphonate derivatives with alkaline phosphatase inhibitory effects: In vitro and docking studies[J]. Journal of Enzyme Inhibition and Medicinal Chemistry, 2018, 33(1): 479-484. DOI:10.1080/14756366.2018.1428193
[13]TRAN H, MCCALLUM T, MORIN M, et al. Homocoupling of iodoarenes and bromoalkanes using photoredox gold catalysis: A light enabled Au(III) reductive elimination[J]. Organic Letters, 2016, 18(17): 4308-4311. DOI:10.1021/acs.orglett.6b02021
[14]MONDAL S, DEBNATH S, DAS B. Synthesis of seven-membered fused sultones by reductive Heck cyclization: An investigation for stereochemistry through DFT study[J]. Tetrahedron, 2015, 71(3): 476-486. DOI:10.1016/j.tet.2014.11.068
[15]ALLEN P, BRAGG R A, CAFFREY M, et al. The synthesis of a tritium, carbon-14, and stable isotope-labeled cathepsin C inhibitors[J]. Journal of Labelled Compounds & Radiopharmaceuticals, 2017, 60(2): 124-129. DOI:10.1002/jlcr.3483
[16]STEINHARDT R C, O'NEILL J M, RATHBUN C M, et al. Design and synthesis of an alkynyl luciferin analogue for bioluminescence imaging[J]. European Journal of Organic Chemistry, 2016, 22(11): 3671-3675. DOI: 10.1002/chem.201503944
[17]LIN K, WILES R J, KELLY C B, et al. Haloselective cross-coupling via Ni/Photoredox dual catalysis[J]. ACS Catalysis, 2017, 7(8): 5129-5133. DOI: 10.1021/acscatal.7b01773
[18]JOSE? A, GARCíA L, MELIHA C?, et al. Synthesis of hindered biaryls via aryne addition and in situ dimerization[J]. Organic Letters, 2015, 17(11): 2649-2651. DOI: 10.1021/acs.orglett.5b01115
[19]WANG Z, LIU Z, LEE W, et al. Design, synthesis and docking study of 5-(substituted benzylidene) thiazolidine-2,4-dione derivatives as inhibitors of protein tyrosine phosphatase 1B[J]. Bioorganic & Medicinal Chemistry Letters, 2014, 24(15): 3337-3340. DOI:10.1016/j.bmcl.2014.05.099
[20]TAKAHASHI S, SUDA Y, NAKAMURA T, et al. Total synthesis of kehokorins A-E, cytotoxic-terphenyls[J]. The Journal of Organic Chemistry, 2017, 82(6): 3159-3166. DOI:10.1021/acs.joc. 7b00147
[21]BRACCA A, KAUFMAN T, CORTéS I, et al. Total synthesis and cytotoxic activity of 6,8-dimethoxy-1,3-dimethylisoquinoline isolated from ancistrocladus tectorius: A 6π-azaelectro-cyclization approach[J]. Synthesis, 2018, 51(2): 433-440. DOI:10.1055/s-0037-1610276
[22]ATTALURI S, IDEN C R, BONALA R R, et al. Total synthesis of the aristolochic acids, their major metabolites, and related compounds[J]. Chemical Research in Toxicology, 2014, 27(7): 1236-1242. DOI: 10.1021/tx500122x
[23]YOSHIDA S, MORITA T, HOSOYA T. Synthesis of diverse benzotriazoles from aryne precursors bearing an azido group via inter- and intramolecular cycloadditions[J]. Chemistry Letters, 2016, 45(7): 726-728. DOI:10.1246/cl.160349
[24]NERVIG C S, WALLER P J, KALYANI D. Palladium-catalyzed intramolecular C-H arylation of arenes using tosylates and mesylates as electrophiles[J]. Organic Letters, 2012, 14(18): 4838-4841. DOI:10.1021/ol302166n
[25]ALI R, GUAN Y, LEVEILLE A N, et al. Synthesis and anticancer activity of structure simplified naturally inspired dimeric chromenone derivatives[J]. European Journal of Organic Chemistry, 2019, 10(41): 6917-6929.
[26]CLARK C G, FLOUDAS G A, LEE Y J, et al.Molecularly tethered amphiphiles as 3-D supramolecular assembly platforms: Unlocking a trapped conformation[J]. Journal of American Chemical Society, 2009, 131(24): 8537-8547. DOI:10.1021/ja900999f
[27]TSUJIYAMA S I, SUZUKI K. Preparation of benzocyclobutenone derivatives based on an efficient generation of benzynes[J]Organic Syntheses, 2007, 84: 272-284. DOI:10.15227/orgsyn.084.0272

Selective synthesis of 2-iodophenyl-3-(-tosyloxy) 4-methylbenzenesulfonate
PAN Feng, WANG Lei, GUO Yi, SHEN Jinying, PAN Xiaofeng, ZHENG Weixin
(311121)
Halogenated sulfonyloxyaromatic ether has been regarded as the polyfunctionalized organic compound with wide applications in lots of fields. Using 2-iodoresorcinol as the starting material, series of iodonated alkyloxy 4-methylbenzenesulfonate were synthesized in high yields via stoichiometrical bissulfonylation, selective monohydrolysis of 2-iodo-1,3-phenylene bis(4-methylbenzenesulfonate) followed by nucleophilic substitution to various organobromides. Procedure for preparation of 3-hydroxy-2-iodophenyl-(4-methylbenzenesulfonate) was explored. It was found that the monodesulfonylation of bissulfonate was much more favorite than monohydrocarbylation for selective functionalization of the two symmetrical hydoxyls in 2-iodoresorcinol. Using low-cost alkali system, the reaction time, temperature of the monohydrolysis of the 2,6-bis(tosyloxy)iodobenzene had been investigated. The monohydrolysis product, 3-hydroxy-2-iodophenyl-(4-methylbenzenesulfonate), were obtained in the purity of above 99% in1H-NMR without further purification. This study provides a synthetic method of 3-alkyloxy-2-iodophenyl 4-methylbenzenesulfonate, which had the advantages of simple operation, mild reaction conditions, low cost and high selectivity. The structures of all the products were verified by1H-NMR,13C-NMR and HRMS, et al.
3-(-tosyloxy)-2-iodophenyl ether; synthesis; selective monohydrolysis
10.3785/j.issn.1008-9497.2021.05.009
O 627
A
1008?9497(2021)05?579?05
2020?01?01.
國家自然科學基金資助項目(20972037).
潘峰(1994—),ORCID: https://orcid.org/0000-0001-7016-2484,男,碩士研究生,主要從事導向金屬有機合成研究,E-mail: pfgz0419@163.com.
,ORCID: https://orcid.org/0000-0003-4149-8100,E-mail:wxzheng@hznu.edu.cn.

