血液樣本蛋白質組分析方法的比較研究
2021-09-14 04:37:16王智博王道平苗蘭李瑛潘映紅劉建勛
生物技術通報 2021年8期
王智博 王道平 苗蘭 李瑛 潘映紅 劉建勛
(1. 中國中醫科學院西苑醫院基礎醫學研究所,北京 100091;2. 中國農業科學院作物科學研究所,北京 100081;3. 北京市中藥藥理學重點實驗室,北京 100091)
蛋白質組學技術作為基因組學和轉錄組學的補充,可用于基因組所表達蛋白的定性定量[1],在發現疾病潛在蛋白分子標志物和藥物靶點以及探究蛋白作用機制等領域具有廣闊的應用前景。蛋白質組學常以細胞、組織和血液為研究材料,其中血液樣本因其微創易得且更能反應生物體的綜合狀態,目前廣泛用于相關研究中[2-4]。
蛋白質組學研究的實驗流程主要由樣本制備、質譜分析、數據處理和生物信息學分析等環節構成。在這些研究環節中,屬于樣本制備范疇的預處理和酶切技術可顯著影響蛋白鑒定率和數據重復性等后續質譜分析結果[5]。盡管當前應用廣泛的超濾法樣本制備技術簡化了溶液內酶切的濃縮和除鹽步驟,能減少樣本損失,獲得較高純度肽段[6],血液樣本的預處理和酶切技術仍需進一步完善。
血液樣品的復雜構成特別是高豐度蛋白是影響蛋白質組學分析效果的主要因素[7]。研究表明基于抗體的親和色譜吸附可有效去除血樣中的高豐度蛋白[8]。基于不同的研究目的,血液樣品可分成血清和血漿,其中血清在制備過程中已去除大部分纖維蛋白原,常用于蛋白質組學分析[9]。酶切技術在血液樣本制備中的重要性也不容忽視。酶切前通常進行蛋白質變性處理,其中熱變性較化學變性與質譜兼容性高,操作簡便且不引入外來干擾物,可作為化學變性的替代方法[10]。Schniers等和Betancourt等[11-12]還報道在低濃度尿素溶液進行胰蛋白酶酶切時,可獲得較好的重復性和深度的蛋白組覆蓋。此外,多數胰蛋白酶切在37℃條件下進行,但Budnik等[13]報道在45℃條件下也可獲得較好的酶切效率。有關酶切技術的優化有大量報道,這些改進技術值得制備血液樣本時參考。
質譜技術是蛋白質組學的關鍵技術。Orbitrap質譜技術是現階蛋白質組分析的主流技術之一,其獲得碎片離子信息的方式常包括數據依賴性采集(data dependent acquisition,DDA)、非數據依賴性 采集(data independent acquisition,DIA)和平行反應監測(parallel reaction monitoring,PRM)3種模式[14]。DDA即經典的鳥槍法,是基于質譜的蛋白質組學實驗的首選方法[15],該模式選擇一級質譜上峰強度相對高的母離子進行碎裂,但母離子單同位素峰不一定是最高峰,低強度峰和共洗脫峰也較難鑒別。DIA解決了離子選擇隨機性的問題,不依賴預設參數,通過固定窗口或可變窗口進行高低離子交替采集,理論上重復性更高,相比DDA在質譜重復上潛力顯著,但數據解析難度大[16]。Venable等[17]將DIA應用于酵母全細胞裂解物分析中,相比DDA-MS直接掃描定量獲得了更高的信噪比、靈敏度、選擇性和動態范圍。近年來DIA譜圖軟件解析軟件不斷更新,極大提升了蛋白質組數據定量的準確度[18]。不同于DIA技術,PRM聚焦于目標肽段產物離子的監測[19]。PRM靶向蛋白質組學技術具有較高的精確度和重復性,但需要預先設定待檢測前體離子,且檢測數量有限[20],因此常應用于特定蛋白驗證。Silva等[21]使用混合消化高密度脂蛋白(HDL)中的標記多肽進行DIA和PRM分析,發現兩種模式均可獲得較好的蛋白定量線性范圍、準確度和精密度。
現有的血樣制備分析方案較多,但發現生物標志物的進度仍舊緩慢[2]。血液樣本包含重要的生物信息,其蛋白質組分析的意義重大。通過血樣蛋白質組分析方法的比較研究,有望建立蛋白鑒定覆蓋率較高和重復性較好的血液樣本制備和分析方法,為進一步的蛋白質組學研究創造條件。
1 材料與方法
1.1 材料
1.1.1 動物 動物實驗選用6周齡SPF級SD大鼠1只,體質量180 g,由北京維通利華實驗動物技術有限公司提供,許可證號SCXK(京)2019-0001。取血前飼養于中國中醫科學院西苑醫院SPF級實驗動物中心,溫度(33±2)℃,相對濕度45%-65%。本研究通過中國中醫科學院西苑醫院動物倫理委員會批準(2018XLC003-2)。
1.1.2 試劑 水合氯醛購自國藥集團化學試劑有限公司(批號30037517);色譜用水購自中國杭州娃哈哈有限公司;尿素(urea)購自美國昂飛公司(Affymetrix);胰蛋白酶(trypsin)購自普洛麥格 公 司(Promega);甲 酸(formic acid,FA)、二硫蘇糖醇(dithiothreitol,DTT)、乙腈(ACN)購自美國賽默飛世爾公司(Thermo Fisher);碘乙酰胺(iodoacetamide,IAA)購自GE公司;碳酸氫銨(ammonium bicarbonate)購自Amresco公司。
1.1.3 儀器與耗材 Easy nLC 1000 納升級液相色譜儀(美國Thermo Fisher公司);Q Exactive Plus 質譜儀(美國Thermo Fisher公司);預柱(富集柱,目錄號HS-Trap-C-5U-5CM)和分析柱(直噴毛細色譜柱,目錄號HS-Anal-C-5U-15CM)購自北京樂潤峰科技有限公司(Beijing Happy Science Scientific Co.Ltd);CS5000型動物體重秤(上海奧豪斯儀器有限公司);ICE-CL31R型低溫離心機(美國Thermo Fisher公司);-80℃超低溫冰箱(美國Thermo Fisher公司);ProteoExtract Albumin/IgG Removal Kit(美國Merck公司,目錄號122642)。
1.2 方法
研究流程見圖1,采用基于超濾膜的過濾輔助法制備樣品。使用維恩圖(https://bioinfogp.cnb.csic.es/tools/venny/)進行定性結果重復性分析。主成分分析和數據可視化使用R軟件包ggplot2_3.3.0和ggfortify_0.4.9。其他數據統計軟件為Excel。
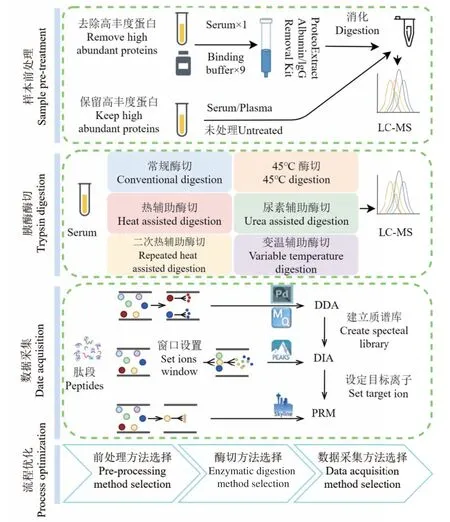
圖1 實驗流程Fig.1 Experimental flow
常規酶切方法、色譜質譜參數設置和數據庫搜索方法參考文獻方法進行[5]并稍作修改。流動相,A為純水,B為CAN,均含0.1% FA(V/V)。流速設定420 nL/min,梯度洗脫設定:0.4 mL/min,3%-6% 3 min,6%-22% 78min,22%-100% 1 min,100% 8min。質譜分析在Q Exactive Plus質譜儀上完成。DDA采集模式的一級質譜分辨率70 000,掃描范圍300-1 800 m/z,自動增益(automated guided cart,AGC)值3e6,注入時間(IT)50 ms。二級質譜的分辨率17 500,AGC值1e5,標準化碰撞能(normalized collision energy,NCE)27.0。毛 細管溫度275℃,電噴霧離子源1900V,二級采集標準為強度前20的母離子進行Orbitrap檢測。質譜原始數據分別采用Proteome Discoverer 2.1(Thermo Fisher) 和 MaxQuant 1.3.0.5(https://www.coxdocs.org/)對原始數據的RAW文件定性和定量,搜索引擎SEQUEST HT。用UniProt Rattus norvegicus 蛋白數據庫(更新時間2020.11.1)進行檢索。設定參數如下,高置信度,肽段長度設定6-144,最大漏切位點為2,前體離子質量容差±10 μmol/L,碎片離子質量容差±0.02 Da,固定修飾設定半胱氨酸碘乙酰胺化(carbamidomethy/+57.021 Da),可變修飾選擇甲硫氨酸氧化(oxidation/+ 15.995 Da)和N-乙酰化(Acetyl/+42.011 Da),蛋白和多肽定性假陽性率FDR為1%。每個樣本的3次質譜重復合并檢索,得到蛋白定性數據以Excel格式導出。
1.2.1 血樣預處理 血清采集:采集全血于試管中,4℃靜置30 min待凝固后離心(1 500×g,10 min),取上清置于EP管中,-80℃保存。血漿采集:采集全血于醫用血常規管(EDTA抗凝)中,4℃離心(1 500×g,10 min),取最上層置于EP管中,-80℃保存。血清去除高豐度蛋白處理:-80℃保存的血清解凍后,參考ProteoExtract Albumin/ IgG Removal Kit說明書去除高豐度蛋白。
3種預處理的大鼠血液樣本按照常規方法進行蛋白組樣品制備和質譜檢測,步驟如下:樣本的初始體積均為50 μL,濃縮后置于超濾管采用8 mol/L尿素洗滌3次,終濃度10 mmol/L DTT還原(37℃,30 min),8 mol/L尿素洗滌3次,終濃度20 mmol/L IAA烷基化(37℃,30 min),8 mol/L尿素洗滌3次,采用50 mmol/L NH4HCO3置換尿素后加入2 μg Trypsin進行酶切(37℃,16 h),離心(14 000 r/min,10 min)取下層液用于質譜分析。利用數據處理軟件比較血漿、血清和去除高豐度蛋白血清的蛋白鑒定數、肽段數、譜圖匹配數和蛋白定量重復性。
1.2.2 胰蛋白酶切(protein digestion) 取50 μL樣本用于酶切處理,還原烷基化步驟同上,8 mol/L尿素和50 mmol/L NH4HCO3洗滌,最終得到約20 μL樣品用于酶切。
1.2.2.1 常規酶切(conventional digestion,CD) 采用常規方法進行還原、烷基化和洗滌,加入2 μg Trypsin,37℃恒溫水浴16 h,加入0.1%(V/V)甲酸水溶液終止酶切,更換接收管,離心(12 000 r/min,20 min),離心液用于質譜分析。
1.2.2.2 45℃孵育(45℃ digestion,45D) 按常規酶切方法處理樣品,孵育溫度設為45℃。
1.2.2.3 熱輔助酶切(heat assisted digestion,HD)樣品還原前加入50 mmol/L NH4HCO3定容到1 mL,在密閉的微型離心管中加熱(95℃,10 min),轉移到冰水浴中降溫終止變性過程,濃縮,余下步驟同CD法。
1.2.2.4 二次熱輔助酶切(repeated heat assisted digestion,RHD) 樣本還原前加入50 mmol/L NH4HCO3定容到1 mL,在密閉的微型離心管中加熱(95℃,10 min),降至室溫后濃縮,加入1 μg酶,37℃孵育3 h,二次加熱樣品(95℃,10 min),降溫后再加入1 μg酶,37℃孵育13 h。
1.2.2.5 尿素輔助酶切(urea assisted digestion,UD) 采用常規方法進行還原、烷基化和洗滌,酶切前加入終濃度為1 mol/L的尿素,余下步驟同CD法。
1.2.2.6 變溫酶切(variable temperature digestion,VD) 采用常規方法進行還原、烷基化和洗滌,酶切前將樣品管置于80℃水浴鍋,待水溫降至60℃后加入含2 μg Trypsin的酶液,隨后轉入37℃水浴鍋孵育16 h。
1.2.3 質譜數據采集
1.2.3.1 數據依賴性采集(DDA) DDA條件設定及數據處理主要參考文獻方法設定[5],其余參數設置見前述。
1.2.3.2 非數據依賴性采集(DIA) 肽段液相分析方法同上,質譜設定為DIA采集模式,一級質譜設定為Full MS,二級質譜掃描設定60個固定掃描窗口,分辨率、掃描范圍、AGC值、注入時間等參數同DDA。將DIA數據導入到PEAKS studio 8.5進行定性和定量,數據采集模式設定為DIA,首先使用DDA采集模式的raw文件構建DIA檢索的蛋白譜圖庫,再對數據進行從頭序列(de novo sequencing) 分析,肽段長度、離子質量偏差、假陽性率、固定修飾和可變修飾等參數同DDA,每個樣本的3次質譜重復合并檢索,得到蛋白定性數據以Excel格式導出。
1.2.3.3 平行反應監測(PRM) 肽段液相分析方法同DDA,質譜設定為PRM采集模式,在設置菜單中選擇離子對設置,將母離子和子離子質量分析儀設置為Orbitrap,分析時間90 min,正離子檢測,MS1 分辨率為70 000(200 m/z),MS/MS 分辨率為17 500(200 m/z),AGC、NCE、IT等參數同DDA。將PRM定量的21個靶蛋白導入Skyline軟件(https://skyline.ms/)中,選擇肽段設置,添加背景蛋白組數據庫文件,根據譜庫中的離子信號選擇蛋白質定量的多肽,從Skyline導出包含保留時間的相關肽列表,并手動檢查靶蛋白的每個肽的定量結果。
1.2.4 血清蛋白組分析流程優化 根據上述血樣蛋白質組分析方法的比較研究結果,采用優化的前處理、酶切和數據采集方法構建適宜的血樣蛋白組分析流程。
2 結果
2.1 血樣前處理方法比較
每個樣品進行3次重復實驗,其中血漿樣本平均檢測到321組蛋白,重復鑒定的蛋白占比66.2%;保留高豐度蛋白血清平均檢測到346組蛋白,重復鑒定的蛋白占比64.4%,相較于血漿增加了16組蛋白鑒定數;去除高豐度蛋白血清平均檢測到377組蛋白,重復鑒定的蛋白占比69.4%,相較于保留高豐度蛋白血清增加31組蛋白鑒定數(圖2-A)。進一步的蛋白定性分析結果顯示,去除和保留高豐度蛋白血清樣品的蛋白鑒定數和二級譜圖數無顯著性差異,但去除高豐度蛋白樣品的肽段和譜圖匹配數(peptide spectrum matches,PSMs)均高于保留高豐度蛋白樣品(圖2-B)。3種血液樣本定量數據的主成分分析顯示,3種血樣的組間分離度明顯較高,但每種血樣的3個重復樣本彼此相近,如圖2-C所示。
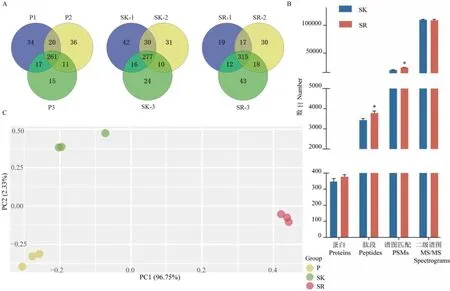
圖2 三種預處理方法制備血樣的質譜結果比較Fig. 2 Comparison of mass spectrometry results of blood samples prepared by 3 pre-processing methods
2.2 酶切方法比較
對不同酶切樣本質譜鑒定的蛋白數、肽段數、譜圖匹配數進行統計,如圖3-A所示,45D組所得蛋白鑒定覆蓋率低于CD組;HD組鑒定到的蛋白數、肽段數和譜圖匹配數較其他組高,其中肽段數和譜圖匹配數相比CD組有顯著性差異,低標準差表明HD組重復性更高;RHD組蛋白鑒定數較CD組低,但識別的肽段數高于CD組;UD組顯示蛋白數、肽段數和譜圖匹配數相對CD組低。而VD法所得蛋白和肽段鑒定率較CD組高,無顯著性差異;另外,VD組各項指標顯示出較高的標準差。分析每組樣品的3次重復制樣,45D組重復率為69.7%,高于其他5組圖;HD組3次重復鑒定到的共有蛋白數最多,為288個(圖3-C)。比較各組樣品的遺漏酶切位點數,VD組酶切位點遺漏率為51.22%,較其他組最低,而RHD組和UD組具有相對較高的漏切率(圖3-B)。另外,分別統計了蛋白序列覆蓋度在20%-35%、35%-50%和>50%的區間分布(圖3-D),發現在20%-35%和>50%的區間,VD組和HD組相比其他4組具有更高的蛋白序列覆蓋度,其中HD組具有更低的標準差,表明HD組蛋白被酶解的更充分;而在區間35%-50%中,UD組和RHD組有最高的肽段分布。對定量結果的主成分分析顯示,組間交叉明顯,其中45D組在主成分1(PC1)的分析中差異最小,RHD組次之;HD組在主成分2(PC2)的分析中差異最小,VD組次之(圖3-E)。
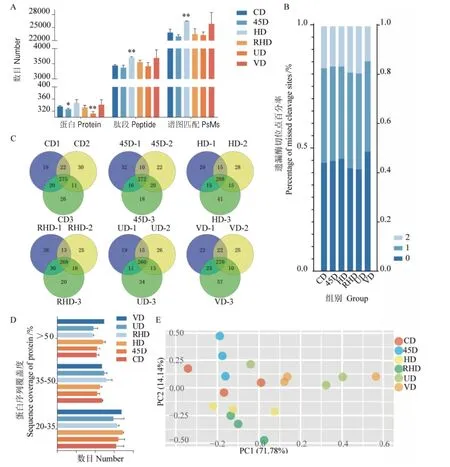
圖3 血清蛋白不同酶切方法質譜結果比較Fig. 3 Comparison of mass spectrometry results of serum proteins with different enzymatic digestion methods
2.3 DDA和DIA數據采集模式結果分析
血清樣品再DDA模式下單次質譜分別鑒定到338、346、359個蛋白;DIA模式下以DDA的raw文件為背景庫單次質譜分別鑒定到371、368、369個蛋白,單次質譜DIA模式更佳(圖4-A)。將3次質譜重復蛋白定性結果合并后剔除重復值,發現DDA和DIA模式分別有422和420個蛋白鑒定數,但DIA具有更小的標準差,且鑒定到的共有蛋白數占總蛋白的比例分別為68.2%和75.9%(圖4-C)。因為質譜分析中普遍存在蛋白數據缺失的狀況,缺失率可作為反映數據采集穩定性和重復性的指標之一,因此根據樣本中蛋白的檢出度,分別統計了DDA和DIA的3次質譜重復蛋白定性數據的缺失值。在DDA模式下,定性數據缺失率分別為26.41%、26.66%、25.12%;在DIA模式下,缺失率分別為11.45%、12.17%、11.93%圖(4-B)。進一步篩選3次質譜重復鑒定到的肽段信息,發現DDA和DIA分別鑒定到共有肽段數為3450和1569,肽段CV值的均數分別為34.65%和22.12%,CV值低于20%的肽段數分別占比29.68%和67.81%圖(4-D)。
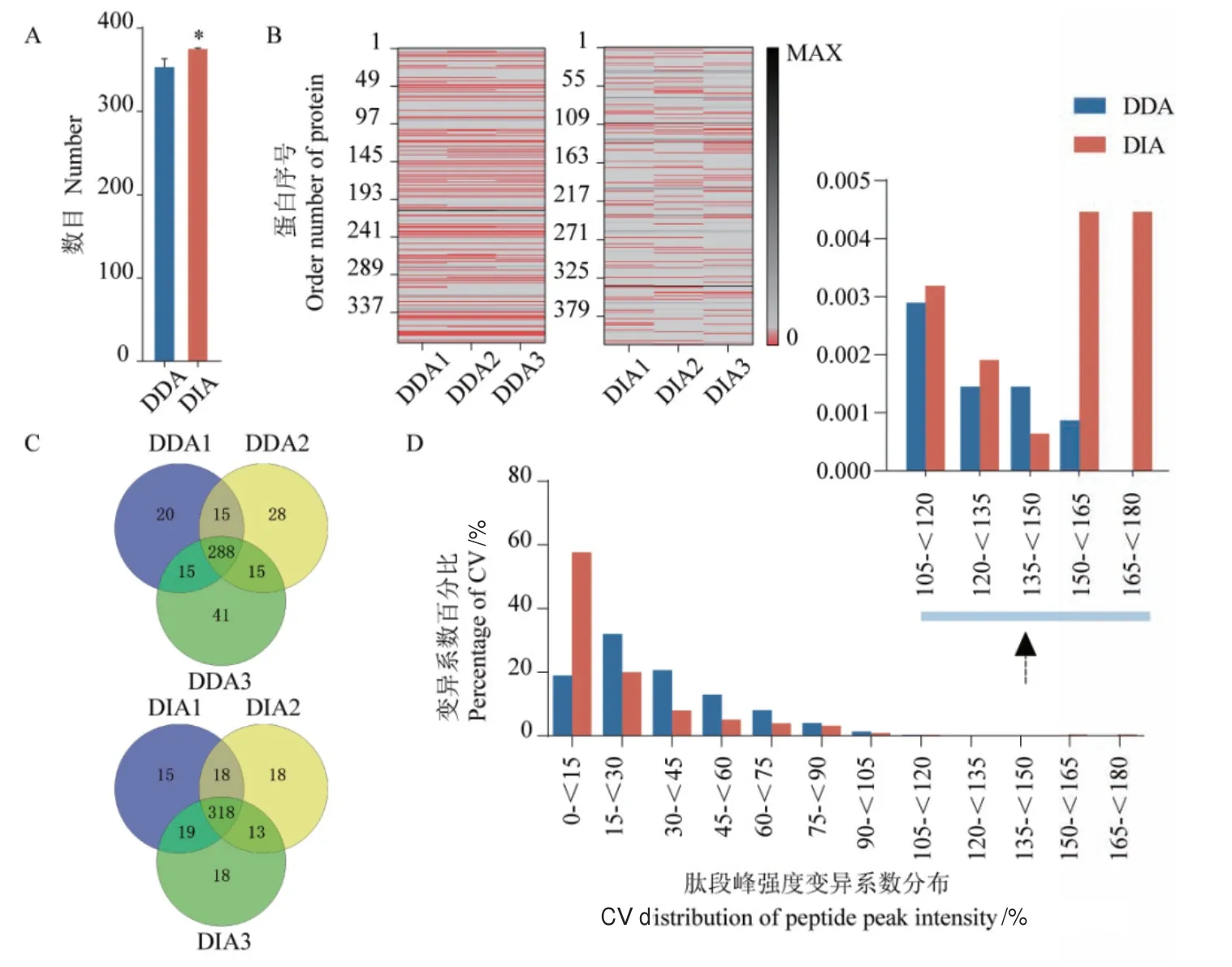
圖4 DDA和DIA數據指標比較Fig. 4 Comparison of DDA and DIA data indices
2.4 DIA和PRM數據采集模式結果比較
選取不同豐度梯度的蛋白比較DIA和PRM在蛋白組中的定量分析能力,通過各特異肽段峰面積計算3次質譜重復的CV值。如表1所示,兩種檢測方法中CV值低于20%的比例分別為42.85%和47.16%,通常篩選特異肽段信息完成蛋白定量,因此多數情況下PRM相對DIA定量更有優勢,在一些蛋白的定量中DIA也展示出良好的定量重復性。
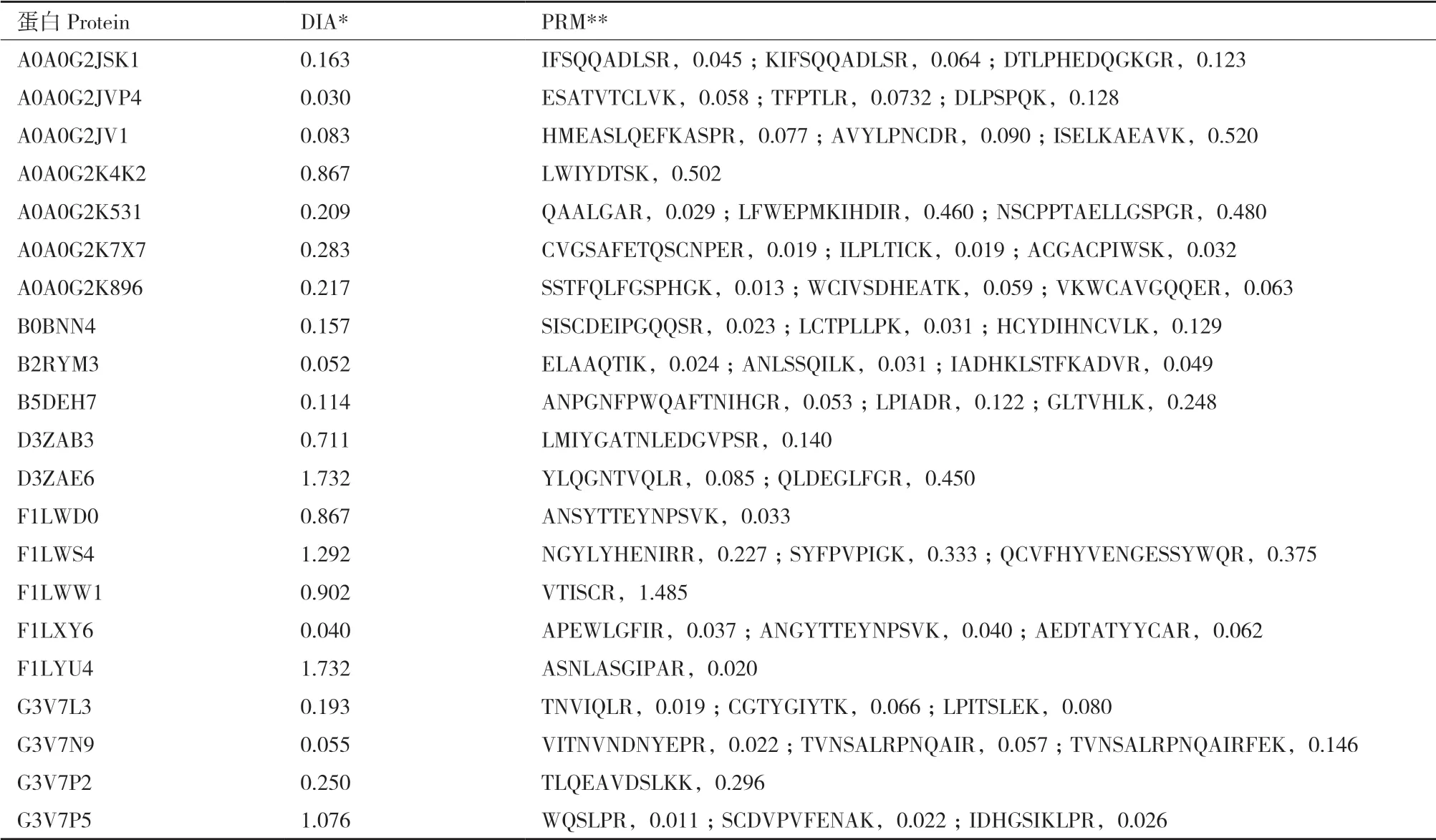
表1 DIA蛋白和對應肽段PRM 3次重復定量CV值分析Table 1 Quantitative analysis on the coefficient of variation of protein from DIA and corresponding peptides from PRM in 3 repeats
2.5 血樣蛋白質組學流程優化
血清去除高豐度蛋白,經熱輔助結合變溫酶切法酶切,最后采用DDA數據采集模式分析,3次重復試驗分別定性到490、490、504個蛋白,鑒定總蛋白數590個,共有蛋白占比69.8%(圖5-A);定量CV值低于20%的蛋白占比71.10%(圖5-B)。
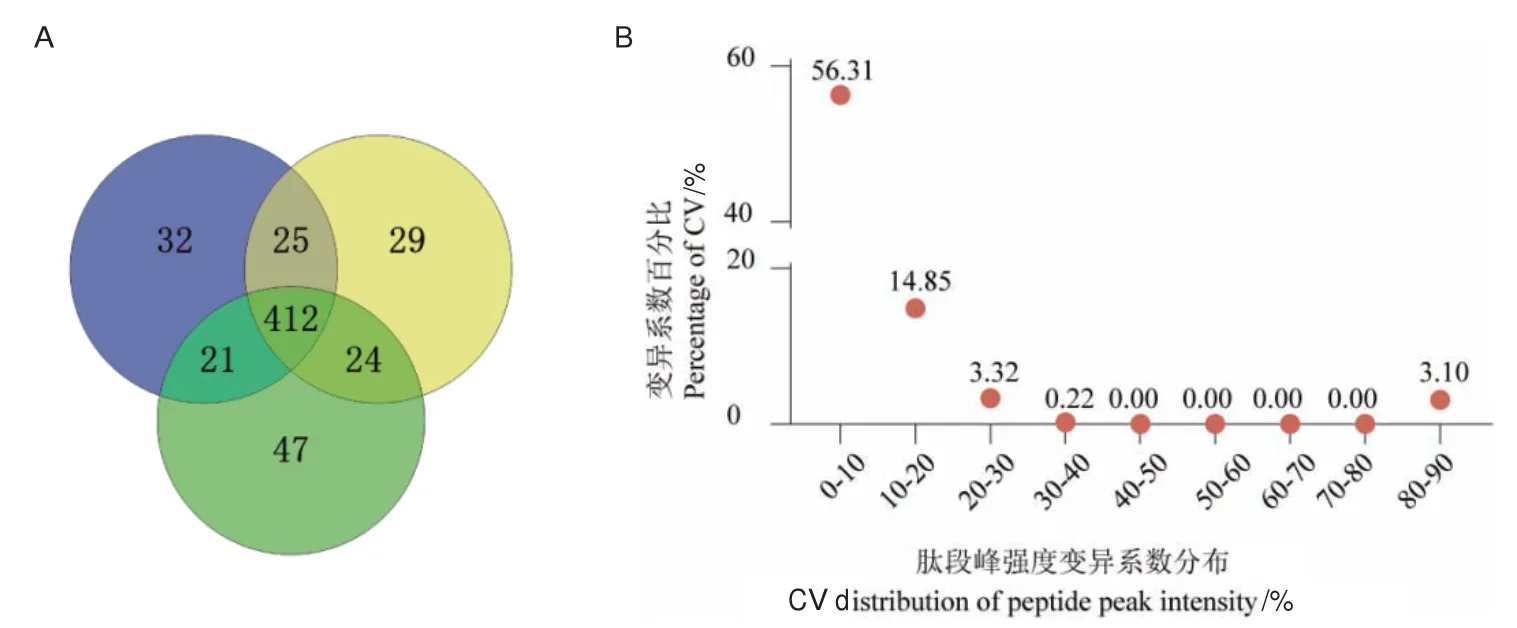
圖5 基于優化流程分析的血樣蛋白組定性和定量結果Fig.5 Qualitative and quantitative results of blood sample proteome based on optimized workflow
3 討論
3.1 樣本前處理結果比較
血樣蛋白質組學研究首要環節是樣本的前處理。定性結果顯示血漿蛋白鑒定數最少,保留高豐度蛋白的血清重復性最低,去除高豐度蛋白的血清蛋白鑒定數和重復性最高,提示血清去除高豐度蛋白可提高蛋白鑒定覆蓋率和可靠性。在相同的色譜質譜條件和數據分析模式下,盡管兩種血清樣品的蛋白鑒定數和二級譜圖數差異不顯著,但去除高豐度蛋白后肽段數和譜圖匹配顯著數增加,證實血清去除高豐度蛋白更適合蛋白質組學研究。定量結果主成分分析顯示血漿和去除高豐度蛋白的血清組內重復性較好,其中去除高豐度蛋白的血清3次重復具有最高的聚合度,表明去除高豐度蛋白處理在血清樣本制備中具有一定的合理性,血清去除高豐度蛋白可獲得更高的蛋白鑒定數和重復率。綜合比較大鼠血樣前處理樣品的質譜鑒定結果,基于研究效率考慮,后續酶切和數據采集方法比較可采用保留高豐度蛋白血清為樣本,但構建優化的血液樣本蛋白質組分析流程時宜采用去除高豐度蛋白血清。
3.2 酶切方法比較
酶切是樣本制備的重要環節,某些蛋白由于存在二硫鍵和特定空間構象,需要通過化學物質或者加熱處理暴露酶切位點,酶切溫度、緩沖液構成等條件同樣影響酶切效率。本研究比較了常規酶切(CD)、45℃孵育(45D)、熱輔助酶切(HD)、二次熱輔助酶切(RHD)、尿素輔助酶切(UD)和變溫酶切(VD)六種方法,發現酶切效率存在差異。HD法鑒定到的蛋白數、肽段數和譜圖匹配數較其他組高,與前人熱變性后肽段鑒定數增加的結果保持一致[10],表明胰蛋白酶酶切前高溫變性更適用于血清樣本蛋白質組分析。胰蛋白酶活性受溫度影響較大,多數文獻在37℃下酶切,但也有45℃的報道[13]。45D法相對CD法在蛋白數、肽段數和譜圖匹配數上較低,說明胰蛋白酶在37℃孵育比45℃酶切效果更佳。RHD相比CD法蛋白數低但肽段數高,原因可能是酶切不完全,從而降低蛋白鑒定率。此外,UD法相對CD法蛋白檢出率低。通常蛋白酶切存在一到多個位點的遺漏酶切,過高的酶切位點遺漏率會降低蛋白定量的精密度和準確度[22]。VD法酶切位點遺漏率最低,表明酶切更充分,但標準差較高,推測原因為在可耐受的范圍內瞬間加入酶液,在溫度變化的窗口區間酶活性激發所致,但實際操作中溫度難以精確控制,從而導致VD法3次重復實驗穩定性差。此外,HD和VD法在在20%-35%和>50%的區間內也顯示出更高的蛋白序列覆蓋度,表明兩種方法得到的血清蛋白質組定性結果更可信和準確。6種酶切方法蛋白定性重復性差異不明顯,其中45D法定性重復率略高,但HD法可獲得更高的共有蛋白數,雖然HD法和VD法的定性重復性不突出,考慮到蛋白鑒定數、漏切位點和蛋白序列覆蓋度,這兩種方法更適用于血清蛋白質
組分析。除了上述基于定性數據的分析外,定量結果的主成分分析顯示45D和RHD組在PC1(71.78%)的分析中差異較小,而HD和VD組在PC2(14.14%)的分析中差異較小,表明CD法和UD法的定量重復性相對較低,45D和RHD更適合血清蛋白質組定量分析。綜合6種酶切方法定性定量結果分析,熱輔助酶切配合變溫酶切更適用血液樣本蛋白質組學研究。
3.3 DDA與DIA比較
3次質譜重復蛋白鑒定數的平均值顯示DIA具有更高的蛋白鑒定數,表明DIA在單次進樣中更有優勢。在蛋白定性數據中,DIA也展示出更好的重復性,合并后剔除重復值顯示DDA和DIA蛋白鑒定數差異不明顯,表明在多次進樣的情況下,兩種方法的蛋白定性能力相差不明顯。缺失值統計結果顯示DIA比DDA平均每組數據完整度高出14.21%,提示DIA具有更高的穩定性和重復性,多數實驗已經證明相同結果[23]。另外,若設定0-15為可信區間,DIA在蛋白定量上顯示出更低的變異系數。因此DIA更適用于篩選血樣中的差異表達蛋白,同時,標記結合靶向定量因其更高的準確度、靈敏度和重現性已廣泛應用于蛋白翻譯后修飾、蛋白互作等領域[24-25]。研究中DIA仍需參考DDA建立的數據庫進行檢索,因此DDA在通量和便捷度上優勢明顯。綜上可得,DIA重復性好且單次質譜鑒定蛋白數高但DDA操作更簡便。
3.4 DIA與PRM比較
DIA和PRM是基于Orbitrap質譜的常用靶向定量方法,選取的21組蛋白或相應肽段CV值顯示,多數特異肽段在PRM定量的CV值低于DIA中對應蛋白定量的CV值,提示PRM具有更高的穩定性,但在實際研究中監測離子數目有限,更適用于候選生物標志物的驗證,DIA和PRM均可用于靶向蛋白質組學分析但PRM結果穩定性相對較高。
3.5 流程優化
去除血樣高豐度蛋白有多種方法,評估各種試劑盒去除高豐度蛋白效率的研究也較多[26-27],本研究采用ProteoExtract Albumin/IgG Removal Kit去除血清高豐度蛋白,可獲得更高的蛋白鑒定數和重復率。常規酶切方法使用廣泛,但也有很多文獻報道優化的酶切方法,如Lys-C/Trypsin順序酶切[28]、微波和紅外輔助酶切[29]等。本研究發現熱輔助酶切配合變溫輔助酶切可獲得更好的質譜結果,更適用于血樣蛋白組研究。在數據采集模式上,DDA和DIA各有優勢,DIA因其較高的重復性和穩定性,單次質譜鑒定蛋白數高,而DDA無需預先建立譜圖庫,可直接通過蛋白序列庫搜索從而完成蛋白的定性定量。PRM定量在多數情況下具有較高的穩定性,但單次進行靶向分析的蛋白數目有限,可用于對目標蛋白的相對和絕對定量。
綜上所述,進行血樣蛋白質組分析時,宜去除高豐度蛋白,采用熱輔助酶切結合變溫酶切法和DDA數據采集模式進行質譜分析,定量結果還可用PRM技術進一步驗證(圖6)。
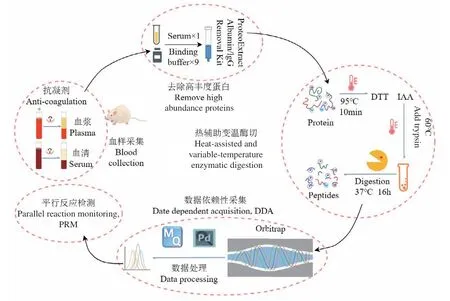
圖6 血樣蛋白質組樣品制備和分析流程Fig. 6 Sample preparation and analysis workflow for blood proteome
4 結論
比較血樣蛋白質組分析方法發現,血清樣本去除高豐度蛋白后,經熱輔助結合變溫酶切法酶切,采用DDA數據采集模式進行質譜分析,可獲得較高的蛋白鑒定覆蓋率和重復性較好的定性定量結果,利用PRM技術還可對定量結果進行后續驗證。基于血液樣本蛋白質組學分析方法的比較研究,建立了優化的制備和分析流程,為進一步的蛋白質組學研究創造條件。
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
