基于RAD-seq簡(jiǎn)化基因組測(cè)序的西藏黃牛遺傳多樣性研究
2021-09-11 01:29:53鄧兵晉美旺杰任增幫次旦歐珠達(dá)瓦羅布楊銳陳仕毅鄺良德
江蘇農(nóng)業(yè)科學(xué) 2021年16期
鄧兵 晉美旺杰 任增幫 次旦歐珠 達(dá)瓦羅布 楊銳 陳仕毅 鄺良德


摘要:為在基因組水平研究西藏黃牛的遺傳多樣性,并通過(guò)選擇信號(hào)分析發(fā)掘重要種質(zhì)特性基因,利用RAD-seq簡(jiǎn)化基因組測(cè)序鑒定拉薩黃牛、阿沛甲咂牛、日喀則駝峰牛和樟木黃牛的SNP標(biāo)記,計(jì)算群體遺傳結(jié)構(gòu)和遺傳進(jìn)化,鑒定基因組受選擇區(qū)域和受選擇基因。結(jié)果表明,共鑒定出1 355 274個(gè)SNP標(biāo)記。阿沛甲咂牛和樟木黃牛遺傳多樣性最為豐富,觀察雜合度分別為0.185 6、0.164 4,核苷酸多樣性分別為0.202 2、0.202 6,拉薩黃牛和日喀則駝峰牛的遺傳多樣性相對(duì)低些。近交系數(shù)最高的為阿沛甲咂牛,而日喀則駝峰牛的近交系數(shù)最低。拉薩黃牛與日喀則駝峰牛之間的遺傳分化系數(shù)最高(0.092 7),而樟木黃牛和阿沛甲咂牛之間的遺傳分化系數(shù)最低(-0.001 1)。聚類分析結(jié)果表明,日喀則駝峰牛和阿沛甲咂牛之間的親緣關(guān)系最近,而與樟木黃牛的親緣關(guān)系最遠(yuǎn)。通過(guò)選擇信號(hào)分析,在西藏黃牛群體中檢測(cè)出22個(gè)基因組區(qū)域受到選擇,包含96個(gè)受選擇基因。GO富集分析表明,這些受選擇基因顯著富集在心血管系統(tǒng)發(fā)育、炎癥反應(yīng)、細(xì)胞間連接、染色質(zhì)結(jié)合等生物學(xué)通路。本研究從基因組水平揭示西藏黃牛的種質(zhì)特性,為進(jìn)一步開展西藏黃牛種質(zhì)資源保護(hù)及利用提供了重要理論依據(jù)。
關(guān)鍵詞:西藏黃牛;遺傳多樣性;簡(jiǎn)化基因組測(cè)序
中圖分類號(hào):S823.8+12 文獻(xiàn)標(biāo)志碼: A 文章編號(hào):1002-1302(2021)16-0153-04
西藏黃牛是西藏的主要地方家畜品種之一,集中分布于西藏農(nóng)區(qū)、半農(nóng)半牧區(qū)和林區(qū),主要分布在海拔 3 000~4 200 m,且分布數(shù)量隨海拔高度的增加而減少。雅魯藏布江中下游、喜馬拉雅山東段和三江流域下游地區(qū)分布較集中,占全區(qū)黃牛總數(shù)的50%以上。西藏黃牛體型外貌以及生產(chǎn)性能由于分布區(qū)域的不同存在一定差異,經(jīng)過(guò)長(zhǎng)期自然選擇形成了拉薩黃牛、阿沛甲咂牛、日喀則駝峰牛、樟木黃牛等4個(gè)主要地方類群。西藏黃牛以產(chǎn)乳為主,乳、肉、役兼用,具有體型小、成熟晚、耐寒、耐粗飼、適應(yīng)高海拔、抗逆性強(qiáng)等特點(diǎn),是我國(guó)寶貴的地方畜禽遺傳資源[1-3]。
通過(guò)利用微衛(wèi)星標(biāo)記和線粒體DNA標(biāo)記來(lái)進(jìn)行遺傳多樣性和遺傳進(jìn)化分析已經(jīng)在不同品種牛上廣泛開展[4-7],然而這些DNA標(biāo)記在整個(gè)牛基因組中的覆蓋范圍是極其微小的,所代表的基因組遺傳變異信息量也是非常有限的。而新一代測(cè)序技術(shù)的出現(xiàn)實(shí)現(xiàn)了SNP分子標(biāo)記的高通量檢測(cè),為從整個(gè)基因組水平上研究牛的遺傳進(jìn)化提供了準(zhǔn)確高效的研究方法[8]。RAD-seq簡(jiǎn)化基因組測(cè)序是通過(guò)限制性內(nèi)切酶對(duì)基因組進(jìn)行打斷,然后進(jìn)行高通量測(cè)序從而得到大量遺傳多態(tài)性標(biāo)簽序列,該技術(shù)流程簡(jiǎn)單、費(fèi)用較低,能有效降低基因組測(cè)序復(fù)雜度,目前,已廣泛應(yīng)用在動(dòng)物分子遺傳標(biāo)記開發(fā)、群體遺傳及進(jìn)化學(xué)、高密度遺傳圖譜構(gòu)建等研究中[9-10]。因此,本研究利用RAD-seq簡(jiǎn)化基因組測(cè)序?qū)ξ鞑攸S牛4個(gè)類群的遺傳多樣性和系統(tǒng)進(jìn)化進(jìn)行研究,能為西藏黃牛遺傳保種提供科學(xué)依據(jù),為進(jìn)一步進(jìn)行品種改良和優(yōu)良新品種培育奠定基礎(chǔ)。
1 材料與方法
1.1 試驗(yàn)動(dòng)物
以西藏黃牛的4個(gè)地方類群,即拉薩黃牛(L)、阿沛甲咂牛(J)、日喀則駝峰牛(T)和樟木黃牛(Z)為研究對(duì)象,于2020年7月在西藏拉薩、林芝、日喀則隨機(jī)采集每個(gè)類群30頭成年牛(公母各半)血液樣本,盡量避免個(gè)體間存在親緣關(guān)系,合計(jì)采集120頭成年西藏黃牛血液樣本,-20 ℃保存?zhèn)溆谩?/p>
1.2 試驗(yàn)方法
1.2.1 RAD-seq簡(jiǎn)化基因組測(cè)序 利用天根血液基因組DNA提取試劑盒(DP318-02)提取樣品基因組DNA,經(jīng)質(zhì)檢合格后,利用Super GBS技術(shù)[1]構(gòu)建pair-end測(cè)序文庫(kù)(300~500 bp),在Illumina PE150平臺(tái)上對(duì)構(gòu)建好的文庫(kù)進(jìn)行RAD-seq簡(jiǎn)化基因組測(cè)序。測(cè)序由上海歐易生物醫(yī)學(xué)科技有限公司完成。
1.2.2 SNP質(zhì)控 測(cè)序結(jié)果用GATK 和Samtools程序進(jìn)行SNP鑒定,質(zhì)控條件為:Q20>95%,ddRAD depth>60%,SNP Call rate>90%,MAF>0.05。
1.2.3 統(tǒng)計(jì)方法 利用Pop Gen 軟件進(jìn)行觀察雜合度(Ho)、近交系數(shù)(Fis)和群體遺傳分化系數(shù)(Fst)計(jì)算,利用Treemix軟件進(jìn)行基因流(Nm)計(jì)算,利用PLINK 軟件進(jìn)行連鎖不平衡(LD)分析和選擇信號(hào)檢測(cè),利用MEGA 軟件UPGMA聚類法進(jìn)行群體聚類分析。
2 結(jié)果與分析
2.1 基因組SNP標(biāo)記鑒定
經(jīng)過(guò)兩步法數(shù)據(jù)質(zhì)控,在西藏黃牛4個(gè)類群中共鑒定出SNP 1 355 274個(gè),其中,常染色體上鑒定出SNP 1 326 666個(gè),性染色體上鑒定出SNP 28 588個(gè),總體上,SNP數(shù)量與染色體長(zhǎng)度呈正相關(guān)。
2.2 西藏黃牛不同類群遺傳多樣性分析
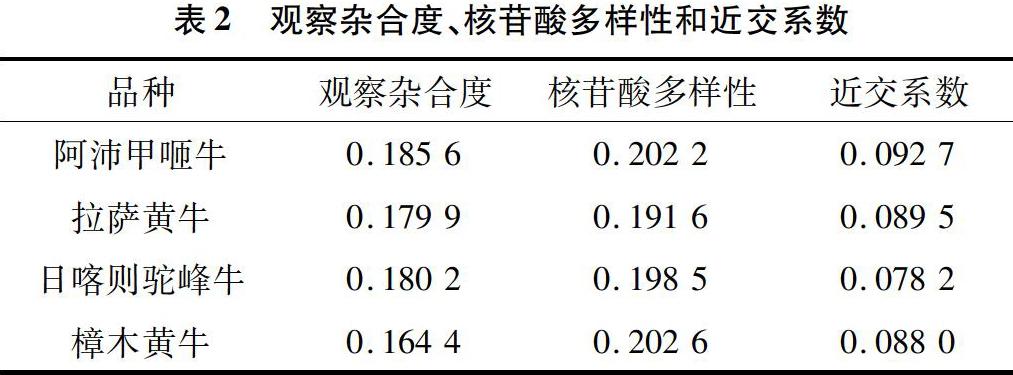
由表2可知,阿沛甲咂牛和樟木黃牛遺傳多樣性最為豐富,觀察雜合度分別為0.185 6、0.164 4,核苷酸多樣性分別為0.202 2、0.202 6,拉薩黃牛和日喀則駝峰牛的遺傳多樣性相對(duì)低些,核苷酸多樣性分別為0.191 6、0.198 5,與LD連鎖不平衡分析結(jié)果(圖1)相一致。近交系數(shù)最高的是阿沛甲咂牛,為0.092 7,日喀則駝峰牛的近交系數(shù)最低,為0.078 2。
2.3 西藏黃牛不同類群遺傳進(jìn)化分析
西藏黃牛不同類群遺傳分化系數(shù)(Fst)和基因流(Nm),由表3可知,拉薩黃牛與日喀則駝峰牛之間的遺傳分化系數(shù)最高(0.092 7),而樟木黃牛和阿沛甲咂牛之間的遺傳分化系數(shù)最低(-0.001 1),相對(duì)應(yīng)的,拉薩黃牛與日喀則駝峰牛之間的基因流最高(0.021 0),而樟木黃牛和阿沛甲咂牛之間的基因流最低(-0.000 3)。UPGMA 聚類結(jié)果,由圖2可知,日喀則駝峰牛和阿沛甲咂牛之間的親緣關(guān)系最近,而與樟木黃牛的親緣關(guān)系最遠(yuǎn)。
2.4 西藏黃牛群體選擇信號(hào)分析
結(jié)合核苷酸多樣性比率(Pi)和遺傳分化系數(shù)(Fst)的選擇信號(hào)分析發(fā)現(xiàn),22個(gè)基因組區(qū)域受到選擇,共有96個(gè)受選擇基因。由圖3可知,功能分析結(jié)果受選擇基因主要富集在心血管系統(tǒng)發(fā)育(VSIG8、TEK、ADM、HOPX)、炎癥反應(yīng)(CCL24、VSIG8、CCL26)、細(xì)胞間連接(TEK、SLAMF8、KDR)、染色質(zhì)結(jié)合(KLF8、MAK、MCMBP、CCNT2)等生物學(xué)通路。
3 討論
3.1 西藏黃牛群體基因組SNP鑒定和遺傳多樣性
本研究通過(guò)簡(jiǎn)化基因組測(cè)序在西藏黃牛4個(gè)地方類群中共鑒定出1 355 274個(gè)SNP位點(diǎn),主要分布在1~19號(hào)常染色體上,且數(shù)量均在40 000個(gè)以上 說(shuō)明西藏黃牛群體具有豐富的遺傳多樣性。本試驗(yàn)克服了微衛(wèi)星標(biāo)記和線粒體DNA技術(shù)在基因組上覆蓋度較低的缺點(diǎn),為從基因組水平進(jìn)行西藏黃牛遺傳進(jìn)化和關(guān)聯(lián)分析奠定了基礎(chǔ),也為構(gòu)建西藏黃牛SNP芯片提供了必要條件。
在西藏黃牛不同類群中,阿沛甲咂牛遺傳多樣性最豐富,觀察雜合度為0.185 6,核苷酸多樣性為0.202 2。同時(shí),其近交系數(shù)最高為0.092 7,提示在阿沛甲咂牛的保種和選種過(guò)程中應(yīng)嚴(yán)格控制近交系數(shù)的過(guò)快增加,維持群體遺傳多樣性的相對(duì)穩(wěn)定。而拉薩黃牛的遺傳多樣性最低,觀察雜合度為0.179 9,核苷酸多樣性為0.191 6。總體來(lái)看這與LD連鎖不平衡分析結(jié)果相一致。拉薩黃牛與日喀則駝峰牛之間的遺傳分化系數(shù)最高(0.092 7),說(shuō)明兩者之間存在中等程度的遺傳分化,而拉薩黃牛與樟木黃牛以及阿沛甲咂牛之間的遺傳分化系數(shù)分別為0.008 5、0.010 1,遺傳分化系數(shù)較小。UPGMA聚類分析表明,日喀則駝峰牛和阿沛甲咂牛之間的親緣關(guān)系最近,而與樟木黃牛的親緣關(guān)系最遠(yuǎn)。總體上,遺傳聚類分析結(jié)果與西藏黃牛4個(gè)地方類群間遺傳分化、基因流以及地理分布相一致。
3.2 西藏黃牛群體選擇信號(hào)
大量研究表明,在動(dòng)物進(jìn)化過(guò)程中,無(wú)論是先天自然選擇,還是后天人工選擇都會(huì)在動(dòng)物基因組上留下選擇信號(hào),受到選擇區(qū)域會(huì)產(chǎn)生高度遺傳分化。因此,進(jìn)行選擇信號(hào)分析有助于進(jìn)一步了解基因組遺傳分化對(duì)動(dòng)物表型的影響,發(fā)掘篩選出影響動(dòng)物性狀的重要的功能基因。目前,國(guó)內(nèi)外學(xué)者利用基因組測(cè)序或基因芯片技術(shù)已經(jīng)在豬、羊、雞等畜禽上開展了選擇信號(hào)研究,發(fā)掘出大量種質(zhì)特性基因和經(jīng)濟(jì)性狀相關(guān)基因[11-14]。針對(duì)牛而言,Zinovieva等利用高密度SNP基因分型芯片對(duì)2個(gè)俄羅斯本地牛品種進(jìn)行選擇信號(hào)研究,檢測(cè)出了新的基因組受選擇區(qū)域和受選擇候選基因[15]。Ben-Jemaa 等利用SNP芯片對(duì)北非牛進(jìn)行基因組選擇信號(hào)研究,確定了36個(gè)受選擇的基因組區(qū)域,篩選到了GH1、ACE、ASIC3、HSPH1等與炎熱相關(guān)的受選擇基因[16]。Mariadassou等利用全基因組測(cè)序?qū)举澟_M(jìn)行選擇信號(hào)研究,找到了57個(gè)受選擇基因組區(qū)域以及68個(gè)受選擇候選基因,其中包括MSTN、NCKAP5、RUNX2等可能與利木贊牛重要表型性狀相關(guān)的基因[17]。Xia等利用全基因組測(cè)序評(píng)估了中國(guó)郟縣紅牛基因組多樣性和選擇信號(hào)特征,采用FST方法找到了17個(gè)受選擇基因,這些基因可能與郟縣紅牛的飼料轉(zhuǎn)化率(CCSER1)、肉品質(zhì)性狀(ROK2、PPP1R12A、CYB5R4、EYA3、PHACTR1)、生育能力(RFX4、SRD5A2)和免疫應(yīng)答(SLAFF1、CD84和SLAFF6)密切相關(guān)[18]。呂世杰等通過(guò)簡(jiǎn)化基因組測(cè)序?qū)χ袊?guó)南陽(yáng)牛和安格斯牛進(jìn)行選擇信號(hào)分析,篩選得到33個(gè)受選擇基因組區(qū)域,其中,16個(gè)基因組區(qū)域與生長(zhǎng)性狀相關(guān)QTLs重合,找到了4個(gè)基因(FXR1、ADAR、IGF1和MNF1)與骨生長(zhǎng)、肌肉發(fā)育和生長(zhǎng)調(diào)控有關(guān)[19]。
在青藏高原上,缺氧是影響動(dòng)物機(jī)體生長(zhǎng)發(fā)育的主要因素,生活在青藏高原上的西藏黃牛具有耐寒、耐粗飼、適應(yīng)高海拔、抗逆性強(qiáng)的特點(diǎn)。本試驗(yàn)中,通過(guò)選擇信號(hào)分析在西藏黃牛群體中發(fā)現(xiàn)22個(gè)基因組區(qū)域受到選擇,找到了VSIG8、TEK、ADM、HOPX等4個(gè)顯著富集在心血管系統(tǒng)發(fā)育的受選擇基因,而心血管系統(tǒng)發(fā)育與動(dòng)物對(duì)高海拔缺氧環(huán)境的適應(yīng)性密切相關(guān)。因此,這4個(gè)受選擇基因極有可能是西藏黃牛對(duì)高海拔適應(yīng)性的候選基因。其中HOPX基因已被證實(shí)在心臟發(fā)育和心肌細(xì)胞增殖中發(fā)揮著重要作用[20-21],而ADM基因參與血管系統(tǒng)調(diào)節(jié)[22],VSIG8和TEK在心血管系統(tǒng)中的作用尚未證實(shí),需要進(jìn)一步研究。同時(shí),與炎癥反應(yīng)相關(guān)的基因CCL24、VSIG8、CCL26也受到了選擇,CCL24和CCL26是趨化性細(xì)胞因子家族成員,能夠促進(jìn)動(dòng)物體內(nèi)炎癥部位的各種白細(xì)胞的補(bǔ)充、激活及黏附[23]。推測(cè)CCL24、VSIG8、CCL26這3個(gè)基因與西藏黃牛的抗逆性強(qiáng)相關(guān),因而在進(jìn)化上受到選擇。
4 結(jié)論
綜上所述,本研究利用簡(jiǎn)化基因組測(cè)序準(zhǔn)確揭示了西藏黃牛4個(gè)類群基因組水平的遺傳多樣性和系統(tǒng)進(jìn)化,選擇信號(hào)分析表明,選擇作用主要集中在高海拔適應(yīng)性、抗逆性等方面的塑造。該研究結(jié)果進(jìn)一步為西藏黃牛種質(zhì)資源的保護(hù)及利用提供了重要參考依據(jù)。
參考文獻(xiàn):
[1]唐建華,陳曉英,宋天增,等. 西藏黃牛種質(zhì)資源保護(hù)與利用研究[J]. 中國(guó)牛業(yè)科學(xué),2016,42(3):48-51.
[2]劉錫武,劉鵬,高玉君. 西藏日喀則市黃牛改良現(xiàn)狀與對(duì)策[J]. 中國(guó)牛業(yè)科學(xué),2009(4):63-65.
[3]石 達(dá). 西藏畜禽品種遺傳資源[M]. 北京:中國(guó)農(nóng)業(yè)大學(xué)出版社,2010.
[4]黃 興,柴志欣,信金偉,等. 西藏牛亞科部分群體線粒體DNA遺傳多樣性研究[J]. 西南民族大學(xué)學(xué)報(bào)(自然科學(xué)版),2019,45(2):117-124.
[5]張桂香,鄭友民,王志剛,等. 我國(guó)部分黃牛品種線粒體D-loop區(qū)遺傳多樣性與起源分化[J]. 遺傳,2009,31(2):160-168.
[6]姬秋梅. 西藏牦牛mtDNA cytb基因的序列多態(tài)性及其系統(tǒng)進(jìn)化分析[J]. 中國(guó)畜牧獸醫(yī)文摘,2013(12):77.
[7]汪 琦,鐘金城,柴志欣,等. 三江黃牛mtDNA Cytb基因序列多態(tài)性及其系統(tǒng)進(jìn)化分析[J]. 中國(guó)畜牧雜志,2016,52(15):20-27.
[8]Davey J W,Hohenlohe P A,Etter P D,et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing[J]. Nature Reviews Genetics,2011,12(7):499-510.
[9]韓 威,朱云芬,殷建玫,等. 基于RAD-seq簡(jiǎn)化基因組測(cè)序的19個(gè)地方雞種遺傳進(jìn)化研究[J]. 畜牧獸醫(yī)學(xué)報(bào),2020,51(4):670-678.
[10]Hohenlohe P A,Amish S J,Catchen J M,et al. Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout[J]. Molecular Ecology Resources,2011,11 (S1):117-122.
[11]Chen Q,Huang Y,Wang Z,et al. Whole-genome resequencing reveals diversity and selective signals in Longlin goat[J]. Gene,2021,771:145371.
[12]Wang M S,Zhang R W,Su L Y,et al. Positive selection rather than relaxation of functional constraint drives the evolution of vision during chicken domestication[J]. Cell Research,2016,26(5):556-573.
[13]Wu F,Sun H,Lu S,et al. Genetic diversity and selection signatures within diannan Small-Ear Pigs revealed by Next-Generation sequencing[J]. Frontiers in Genetics,2020,11:733.
[14]Wang X,Zhang H,Huang M,et al. Whole-genome SNP markers reveal conservation status,signatures of selection,and introgression in Chinese Laiwu pigs[J]. Evolutionary Applications,2021,14(2):383-398.
[15]Zinovieva N A,Dotsev A V,Sermyagin A A,et al. Selection signatures in two oldest Russian native cattle breeds revealed using high-density single nucleotide polymorphism analysis[J]. PLoS One,2020,15(11):e0242200.
[16]Ben-Jemaa S,Mastrangelo S,Lee S H,et al. Genome-wide scan for selection signatures reveals novel insights into the adaptive capacity in local North African cattle[J]. Scientific Reports,2020,10(1):19466.
[17]Mariadassou M,Ramayo-Caldas Y,Charles M,et al. Detection of selection signatures in Limousin cattle using whole-genome resequencing[J]. Animal Genetics,2020,51(5):815-819.
[18]Xia X,Zhang S,Zhang H,et al. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data[J]. BMC Genomics,2021,22(1):43.
[19]呂世杰,陳付英,金 磊,等. 利用簡(jiǎn)化基因組測(cè)序篩選安格斯牛生長(zhǎng)相關(guān)的受選擇基因[J]. 畜牧獸醫(yī)學(xué)報(bào),2020,51(4):713-721.
[20]Chen F,Kook H,Milewski R,et al. Hop is an unusual homeobox gene that modulates cardiac development[J]. Cell,2002,110(6):713-723.
[21]Trivedi C M,Zhu W,Wang Q,et al. Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation[J]. Developmental Cell,2010,19(3):450-459.
[22]史洪巖,賀 綦,程 敏,等. HOPX基因過(guò)表達(dá)對(duì)雞前脂肪細(xì)胞增殖的影響[J]. 中國(guó)農(nóng)業(yè)科學(xué),2015,48(8):1624-1631.
[23]高云飛,余艷紅,李志琴 . ADM基因在正常孕婦與子癇前期孕婦妊娠晚期胎盤組織表達(dá)的研究[J]. 南方醫(yī)科大學(xué)學(xué)報(bào),2006,26(12):1828-1830.顏世卿,馬 軻,陳 巍. 雞腸源益生口乳桿菌的分離鑒定與生物學(xué)特性[J]. 江蘇農(nóng)業(yè)科學(xué),2021,49(16):157-161.
