基因組預(yù)測的序列數(shù)據(jù)在家畜育種中的應(yīng)用
2021-09-05 20:09:43張配配校黃選洋
國外畜牧學(xué)·豬與禽 2021年4期
張配配 校 黃選洋 譯
摘? 要:全基因組序列數(shù)據(jù)的使用在家畜育種計劃中具有巨大的潛力,可以提高發(fā)現(xiàn)變異基因的能力,同時能更準(zhǔn)確和更持久地預(yù)測育種值而不是標(biāo)記陣列。要了解家畜基因組序列數(shù)據(jù)的全部潛力,需要從大量的個體,甚至要從數(shù)百萬個個體上獲得基因組序列和表型數(shù)據(jù),從而準(zhǔn)確地估測構(gòu)成數(shù)量性狀基礎(chǔ)的大量致病變異的影響。
關(guān)鍵詞:全基因組;標(biāo)記陣列;估測;變異;雜交剝離
中圖分類號:S813.3 文獻(xiàn)標(biāo)志碼:C 文章編號:1001-0769(2021)04-0012-03
低成本的測序策略結(jié)合估測法(imputation),能夠以負(fù)擔(dān)得起的成本為大量個體生成所需基因組序列的信息。低覆蓋率使研究人員對大量個體進(jìn)行基因組測序成為可能,這可以提高變異的發(fā)現(xiàn)率,特別是低頻率的變異,并能加強(qiáng)根據(jù)基因組序列數(shù)據(jù)對整個群體的估測。
本文介紹了我們在一項研究中所采用的策略,該研究對來自9個商業(yè)品系的7 848頭豬進(jìn)行了全基因組測序,這些品系大部分處于低覆蓋率范圍。隨后,我們證明,將該測序策略與“雜交剝離”估測法相結(jié)合,是一種可為大群家畜純種系譜產(chǎn)生全基因組序列數(shù)據(jù)的有效策略。最后,我們測試了這些大數(shù)據(jù)集對合成表型的基因組預(yù)測的優(yōu)勢。
1? 材料和方法
1.1 測序策略
我們對Genus plc公司的9個商業(yè)品系(PIC豬商業(yè)品系,公司位于美國田納西州亨德森縣)的7 848頭豬的全基因組進(jìn)行了測序。測序時,我們從每個品系中選擇約2%(1.7%~2.5%)的豬。結(jié)果表明,大多數(shù)豬處于低覆蓋率,目標(biāo)覆蓋率為1倍或2倍,一小部分豬處于較高的覆蓋率,分別為5倍、 15倍或30倍。個體的平均覆蓋率為4.1倍,但中位數(shù)為1.5倍。我們使用三步策略選擇個體和這些個體的覆蓋范圍:
第一步:在純種系譜中貢獻(xiàn)最多基因型后代的父系和母系分別擁有2倍和1倍的覆蓋率。
第二步:AlphaSeqOpt法第1部分用于識別在種群單倍型中占有最大比例的單倍型個體,并在控制總成本的前提下,為它們及其祖先分配一個介于0倍至30倍的最優(yōu)水平的測序覆蓋。
第三步:AlphaSeqOpt法第2部分用于識別累計覆蓋率低(低于10倍)的單倍型個體,并對這些個體進(jìn)行1倍測序,以增加單倍型的累計覆蓋率(即大于或等于10倍)。
AlphaSeqOpt法使用根據(jù)階段性標(biāo)記陣列基因型推斷的單倍型。
1.2 發(fā)現(xiàn)變異
將測序結(jié)果與Sscrofa 11.1參考基因組進(jìn)行比對,利用一個基于GATK 3.8的Haplotype-Caller工具的數(shù)據(jù)來源找出變異。為了避免在應(yīng)用低覆蓋率序列數(shù)據(jù)時對GATK引入的參考等位基因產(chǎn)生誤差,我們利用堆積函數(shù)提取了支持該等位基因的讀取數(shù),結(jié)果從這9個品系中共發(fā)現(xiàn)了6 000萬個單核苷酸多態(tài)性(Single Nucleotide Polymorphisms,SNPs)。
1.3 估測全基因組序列數(shù)據(jù)
使用商業(yè)標(biāo)記陣列對每個群體中的大多數(shù)個體進(jìn)行基因分型,擁有15 000個低密度(Low Density,LD)或75 000個高密度(High Density,HD)全基因組標(biāo)記。正如用AlphaPeel法測算的那樣,采用雜交剝離估測法分別估測每個群體的全基因組序列。該方法通過兩階段,降低估測成本:
· 多軌跡迭代剝離,可以根據(jù)數(shù)組中的該標(biāo)記估計分離概率。
· 改進(jìn)的單位點迭代剝離,可以基于序列數(shù)據(jù)旁側(cè)數(shù)組的該標(biāo)記的估測值,利用該序列數(shù)據(jù)大致估計任何其他變異位點上的分離概率。由于每條染色體中重組基因的數(shù)量有限,以及附近標(biāo)記共同被遺傳的概率很高,這種大致估測的精度損失可以忽略不計。9個品系估測出的豬總數(shù)約為35萬頭。
為了評估估測的準(zhǔn)確性,我們使用了來自4個大小不同的群體在高覆蓋率(15倍或30倍)下測序的284個個體。被檢測個體的序列數(shù)據(jù)用留一法設(shè)計(leave-one-out design)可以完全掩蓋。將估測的等位基因劑量與獲得完整數(shù)據(jù)的等位基因劑量進(jìn)行比較,認(rèn)為是“真”值。
1.4 基因組的預(yù)測
我們在一個擁有3萬個個體的品系中檢測了基因組預(yù)測的準(zhǔn)確性,這些個體的估測基因型為1 600萬個SNPs。正如在AlphaBayes軟件中預(yù)測的那樣,使用嶺回歸( ridge regression)模型預(yù)測基因組。
利用該模式測試了22 318個個體,驗證了1 458個個體。對9個具有不同遺傳力和數(shù)量性狀核苷酸(Quantitative Trait Nucleotides,QTN)的合成性狀進(jìn)行基因組預(yù)測。
使用4組標(biāo)記進(jìn)行基因組預(yù)測:從陣列中預(yù)選5.7萬個標(biāo)記(HD),從基于LD修剪的序列數(shù)據(jù)中預(yù)選24.8萬個變體[全基因組測序(Whole Genome Sequencing,WGS)_LD,WGS_LD],從基于單標(biāo)記回歸結(jié)果[(WGS_基于總數(shù)據(jù)的孟德爾隨機(jī)化(Summary data-based Mendelian Randomization,SMR),WGS_SMR]的序列數(shù)據(jù)中預(yù)選18.3萬個變體,或通過僅每保留第200個變體(WGS_200)從該序列數(shù)據(jù)中預(yù)選6.7萬個變體。基因組估計育種值(Genomic Estimated Breeding Value,gEBV)的準(zhǔn)確性是根據(jù)該驗證數(shù)據(jù)集中g(shù)EBV與合成表型之間的相關(guān)性來估計的。
2? 結(jié)果和討論
2.1 估測的準(zhǔn)確性
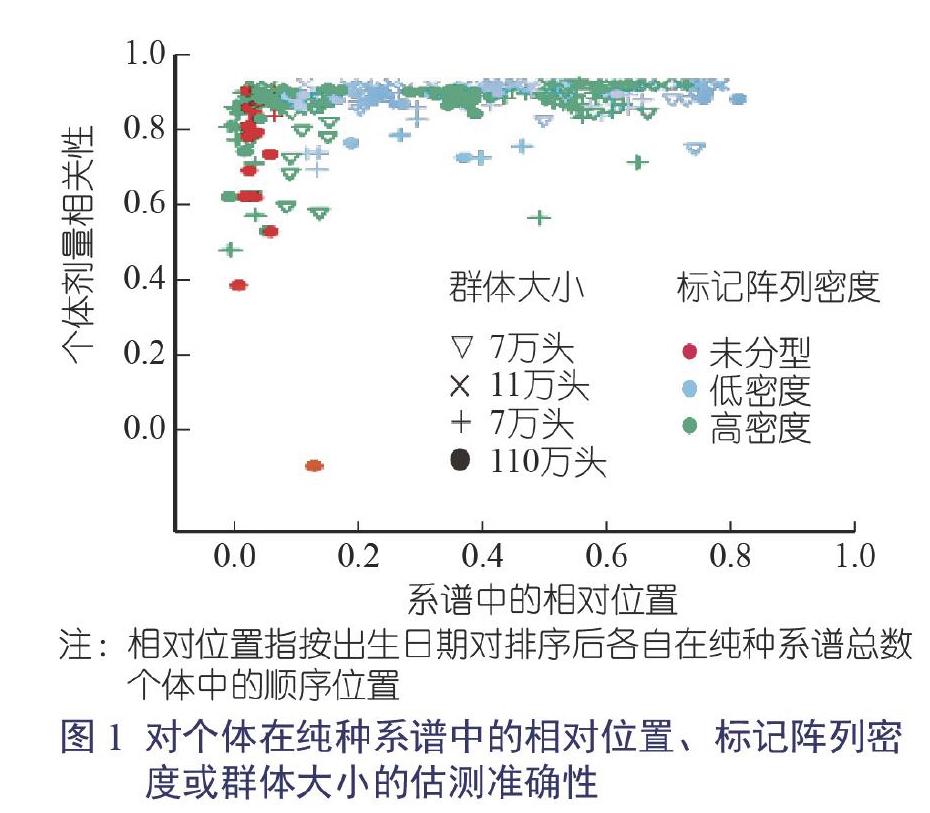
對大多數(shù)受試個體而言,真實數(shù)據(jù)的估測精度較高(圖1)。平均個體劑量相關(guān)性為0.94,中位數(shù)為0.97,四分位數(shù)范圍為0.94~0.98。一些屬于該純種系譜最早幾個世代的最古老的個體(位于系譜的前20%)具有很低的估測精度,因為它們無法提供其直系祖先的信息,或能夠提供的信息極少,這影響了估測精度。
較晚幾個世代的個體(位于系譜中前20%的后面)有更高的估測精度,平均劑量相關(guān)性為0.97,變異性更低:中位數(shù)為0.98,四分位數(shù)間距為0.96~0.99。
個體的標(biāo)記陣列密度與用標(biāo)記陣列基因分型獲得的直系祖先的數(shù)量相矛盾,但對稍后幾個世代的個體而言,標(biāo)記陣列密度的HD和LD之間無顯著差異,種群大小對估測精度的影響無明顯的傾向性。
2.2 基因預(yù)測
在某些情況下,與標(biāo)記陣列相比,序列數(shù)據(jù)能夠提供更好的預(yù)測精度,但其優(yōu)勢取決于該性狀的遺傳結(jié)構(gòu)。
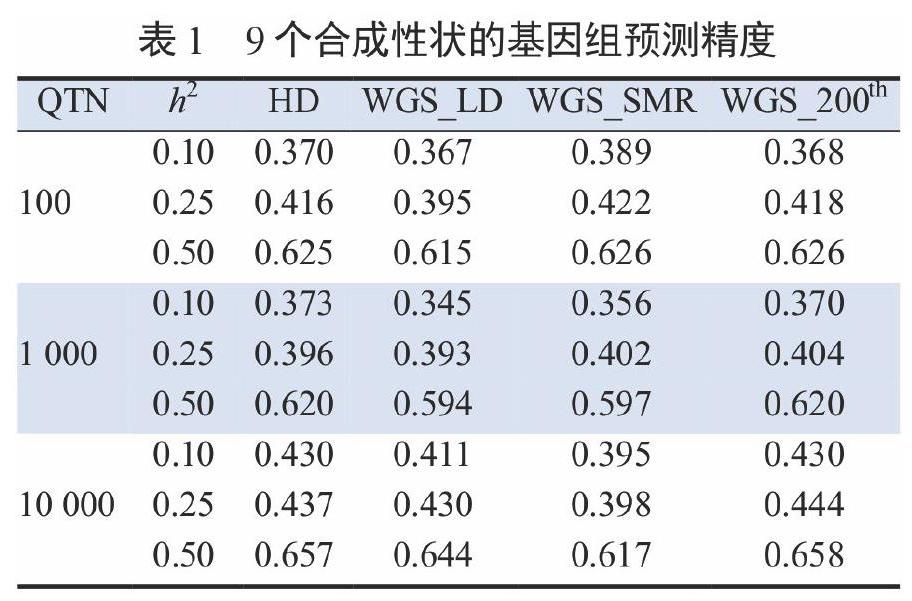
表1列出了9個合成性狀的基因組預(yù)測精度。當(dāng)QTN的數(shù)量較小時,可以識別能支撐該性狀的遺傳變異的變體(variants)具有足夠的統(tǒng)計功效(statistical power),使用這些變體(WGS_SMR)進(jìn)行預(yù)測的準(zhǔn)確性高于用來自商業(yè)標(biāo)記陣列(HD)的標(biāo)記進(jìn)行預(yù)測的。這與之前的觀察結(jié)果一致,添加一個或幾個具有較大作用的標(biāo)記作為預(yù)測因子可以提高該標(biāo)記序列的預(yù)測精度。
當(dāng)QTN的數(shù)量較大時,WGS_SMR的性能比HD的差。在這種情況下,從序列數(shù)據(jù)中選擇的其他變異集可能(略微)比商業(yè)標(biāo)記序列更有利,因為它們不會像商業(yè)標(biāo)記序列那樣受到確定偏倚(ascertainment bias)的影響。
這些結(jié)果部分是由于目前使用商業(yè)標(biāo)記陣列進(jìn)行基因組選擇已經(jīng)獲得了很高的預(yù)測準(zhǔn)確性,且與其他研究結(jié)果一致。后者發(fā)現(xiàn),與HD標(biāo)記陣列相比,序列數(shù)據(jù)在基因組預(yù)測上沒有改善或只有微小的變化。有待確定的是,結(jié)果是否會因以下原因而得到改善:來自多個品種的數(shù)據(jù),使用多品種測試和更大的測試集,或比嶺回歸更適合于大規(guī)模開發(fā)序列數(shù)據(jù)的基因組預(yù)測方法。
3? 結(jié)論
無論種群的規(guī)模多大,只要個體與具有標(biāo)記陣列或序列數(shù)據(jù)的親緣聯(lián)系在一起,同時該親緣有足夠多的信息,恰當(dāng)?shù)臏y序策略和“雜交剝離”的結(jié)合是在大群的純種系譜中生成全基因組序列數(shù)據(jù)的一種有效方法。
目前尚不清楚,這些帶有估測序列數(shù)據(jù)的大數(shù)據(jù)集是否能夠提高基因組預(yù)測的準(zhǔn)確性。
原題名:Sequence data for genomic prediction in livestock breeding(英文)
原作者:Roger Ros-Freixedes等(愛丁堡大學(xué))
