吹掃捕集-氣相色譜-質譜法快速測定同序列地下水和土壤中的37種揮發性有機物
2021-08-29 14:17:26張利鈞楊豐春
理化檢驗-化學分冊 2021年8期
關鍵詞:方法
付 瑤,張利鈞,楊豐春,李 欣,任 偉
(濟南市環境研究院,濟南 250100)
揮發性有機物(VOCs)主要包括芳烴、鹵代芳烴、鹵代烷烴、烯烴類等,因其沸點低、分布廣和“三致”(致癌、致畸、致突變)作用,嚴重危害人類健康和生態環境[1],被列為優先控制污染物。環境中的土壤和地下水關系緊密、相互作用,在實際工作過程中常會出現同時測定兩者中相同指標的情況,且土壤和地下水污染均具有過程緩慢、不易發現、隱蔽性強和難以修復等特點,因此及時檢測進而加強監管修復尤為重要。
目前檢測土壤和地下水中VOCs含量的前處理方法主要包括吹掃捕集法[2-10]、頂空法[11-13]和頂空-固相萃取法[14]等。吹掃捕集作為一種動態頂空技術,可減少傳統頂空法平衡溶劑帶來的二次污染。氣相色譜法常用的檢測器有氫火焰離子化檢測器(FID)[7,12-13]、光離子化檢測器(PID)[15]。FID 無法定性區分色譜峰重疊時VOCs的種類,或是在基體組分復雜時呈現假陽性;PID 僅針對VOCs總量進行測定,且易受土壤含水率和空氣的影響[15],兩者均具有一定的局限性。氣相色譜儀與質譜儀[4-8,13-14]聯用,可快速、靈敏地對VOCs各組分進行定性分析和定量測定。
目前文獻報道主要針對單一地下水或土壤的測試條件進行研究,而對于校準曲線的繪制、吹掃捕集條件優化等試驗過程中常遇到的細節問題較少涉及。國內現行標準HJ 605-2011?土壤和沉積物揮發性有機物的測定 吹掃捕集/氣相色譜-質譜法?及HJ 639-2012?水質 揮發性有機物的測定 吹掃捕集/氣相色譜-質譜法?提供的吹掃捕集參數主要針對舊式冷阱,與應用于當前新式冷阱的參數相比有很大差異。
本工作根據相關質量標準要求[16-17],分別優化了地下水和土壤的校準曲線的繪制、吹掃捕集和氣相色譜-質譜(GC-MS)條件,并對濟南市7個水源地的土壤及地下水樣品進行分析,以期為地下水和土壤的聯合修復提供科學依據。
1 試驗部分
1.1 儀器與試劑
島津QP2020NX 型氣相色譜-質譜聯用儀;Tekmar ATOMX-XYZ型全自動固液一體吹掃捕集裝置;MS205DU 型電子天平;CNW 40 mL 棕色吹掃瓶。
37種VOCs混合標準溶液:2.0 g·L-1,使用前用甲醇稀釋至20 mg·L-1,于-20 ℃保存。
內標溶液:2.0 g·L-1,含氟苯、4-溴氟苯、1,2-二氯苯-d4等3種組分,于-20 ℃保存。
替代物標準溶液:250 mg·L-1,含二溴氟甲烷、甲苯-d8兩種組分,于-20 ℃保存。
抗壞血酸為優級純;甲醇為農殘級;石英砂為分析純;試驗用水為超純水(電導率18.2 MΩ·cm)。
1.2 儀器工作條件
1)吹掃捕集條件 吹掃流量40 mL·min-1,吹掃溫度20 ℃,吹掃時間11 min;干吹時間,測定土壤樣品時需2 min,測定地下水樣品時需0.5 min;預脫附溫度245 ℃,脫附溫度250 ℃,脫附時間1.7 min;烘烤溫度280 ℃,烘烤時間8 min;傳輸線溫度180 ℃。
2)色譜條件 SHIMADZU SH-Rxi-624Sil MS色譜柱(60 m×0.32 mm,0.18μm);進樣口溫度200℃;載氣為氦氣(純度大于99.999%);分流進樣,分流比30∶1;柱流量1.5 mL·min-1;進樣量1.0 μL。柱升溫程序:初始溫度為38 ℃,保持1.8 min;以10 ℃·min-1速率升溫至120 ℃;再以15 ℃·min-1速率升溫至240 ℃,保持2 min。
3)質譜條件 電子轟擊離子源(EI);電離能量70 e V;離子源溫度200℃,傳輸線溫度280℃;選擇離子監測(SIM)模式。其他質譜參數見表1。
1.3 試驗方法
1.3.1 樣品的采集、保存及測定
水樣采樣前,在吹掃瓶內加入25 mg 抗壞血酸。采集樣品時,應盡量避免或減少樣品在空氣中的暴露,并使水樣在瓶口形成上彎月面,不留氣泡,冷藏,于14 d內測定。土壤樣品采樣前,在棕色吹掃瓶中放入一個清潔的磁力攪拌棒,采樣前后密封并稱重(精確至0.01 g),采樣質量約5 g,冷藏,于7 d內測定。
樣品經冷藏后取出,水樣滿瓶,用微量進樣針移出相應體積的水樣后,加入40μg·L-1內標溶液40μL;土壤樣品加入10.00 mL水及40μg·L-1內標溶液10.00μL。再分別加入一定體積的替代物標準溶液,經渦旋儀振蕩混勻后放入樣品架,按儀器工作條件進行測定。
1.3.2 繪制校準曲線的混合標準溶液系列的配制
將適量的37 種VOCs混合標準溶液、替代物標準溶液、內標溶液用水稀釋,并定容至50 mL 容量瓶中,配制成目標物和替代物質量濃度為1,2,4,8,16,32,64,80μg·L-1,內標質量濃度為40μg·L-1的混合標準溶液系列。其中,土壤樣品繪制校準曲線時,取10.00 mL 上述混合標準溶液系列至吹掃捕集樣品瓶中(內置5 g石英砂空白,實際目標物的質量濃度為2,4,8,16,32,64,128,160μg·kg-1);水樣品繪制校準曲線時,取滿瓶(約40 mL)。進樣時,儀器用氣密性注射器自動吸取混合標準溶液5.0 mL,按儀器工作條件測定,內標法定量。
2 結果與討論
2.1 校準曲線繪制條件的優化
繪制校準曲線是多組分易揮發目標物的難點,特別是對于低沸點化合物,操作步驟對相對響應因子(RRF,為目標物響應值與內標響應值的比值與內標質量濃度與目標物質量濃度比值的乘積)及相關系數(r)的影響較大(圖1)。土壤樣品的混合標準溶液系列的優化首先選取兩種配制方法:1#在吹掃瓶內加入5.00 mL 水后,用微量進樣針加入混合標準溶液;2#采用50 mL 玻璃容量瓶配制,移液管移取5.00 mL混合標準溶液至吹掃瓶中。方法1#減少了配制過程中的損失,目標物的RRF 明顯增大,RRF的相
對標準偏差(RSD,n=8)大部分低于20%,但由于存在系統誤差(混合標準溶液系列各點實際的質量濃度不同程度地大于理論質量濃度),部分目標物RRF的RSD(n=8)在20%左右,且校準曲線的相關系數偏低。方法2#校準曲線的相關系數不小于0.990 0,線性良好,但RRF 較低,導致其RSD(n=8)普遍不低于20%,主要原因可能是由于吹掃針在液面上方吹掃不完全。綜合考慮,試驗選擇采用50 mL容量瓶配制后再轉移至吹掃瓶中。
試驗進一步考察了3種進樣方法:3#移液管移取20.00 mL 混合標準溶液至吹掃瓶中;4#移液管移取5.00 mL 混合標準溶液后,吹掃瓶中再加入5.00 mL水;5#移液管移取10.00 mL 混合標準溶液至吹掃瓶中。其中方法3#和5#相比,方法3#部分目標物測定效果較差,這是因為方法3#加入混合標準溶液體積較大,造成吹掃不完全;而方法4#和5#相比,吹掃體積相同,均為10.00 mL,得到的校準曲線經比較,方法5#RRF的RSD(n=8)顯著低于方法4#的,這是因為方法5#中加入的混合標準溶液更為均勻,且一次性加入,避免了二次加水引入污染的可能;方法5#與方法2#相比,90%以上目標物的相關系數不小于0.999 0,僅少量在0.998 0~0.999 0內,線性良好,除容易受環境影響的四氯化碳等少數目標物RRF的RSD(n=8)在5.0%~10%內,其余均小于5.0%,說明在液面以下通氮氣吹掃比液面以上效率更高。因此,試驗選擇方法5#進行校準曲線的繪制。
2.2 吹掃時間的優化
在其他條件相同的情況下,考察了吹掃時間(9,11,13,15 min)對氯乙烯、萘回收率的影響,結果如圖2所示。
由圖2 可知:當吹掃時間由9 min 延長至11 min時,氯乙烯、萘的回收率呈現增高趨勢;當吹掃時間由11 min延長至15 min時,氯乙烯的回收率逐漸降低,而萘的回收率繼續增高且超于穩定,在13 min后緩慢降低。這可能是由于吹掃時間過長,目標物在未加熱脫附前發生部分解吸,被載氣帶出,導致回收率降低。綜合考慮地下水樣品和土壤樣品,并兼顧不同目標物回收率,11 min均是一個較好的平衡點[3-6,9-10]。因此,試驗選擇吹掃時間為11 min。
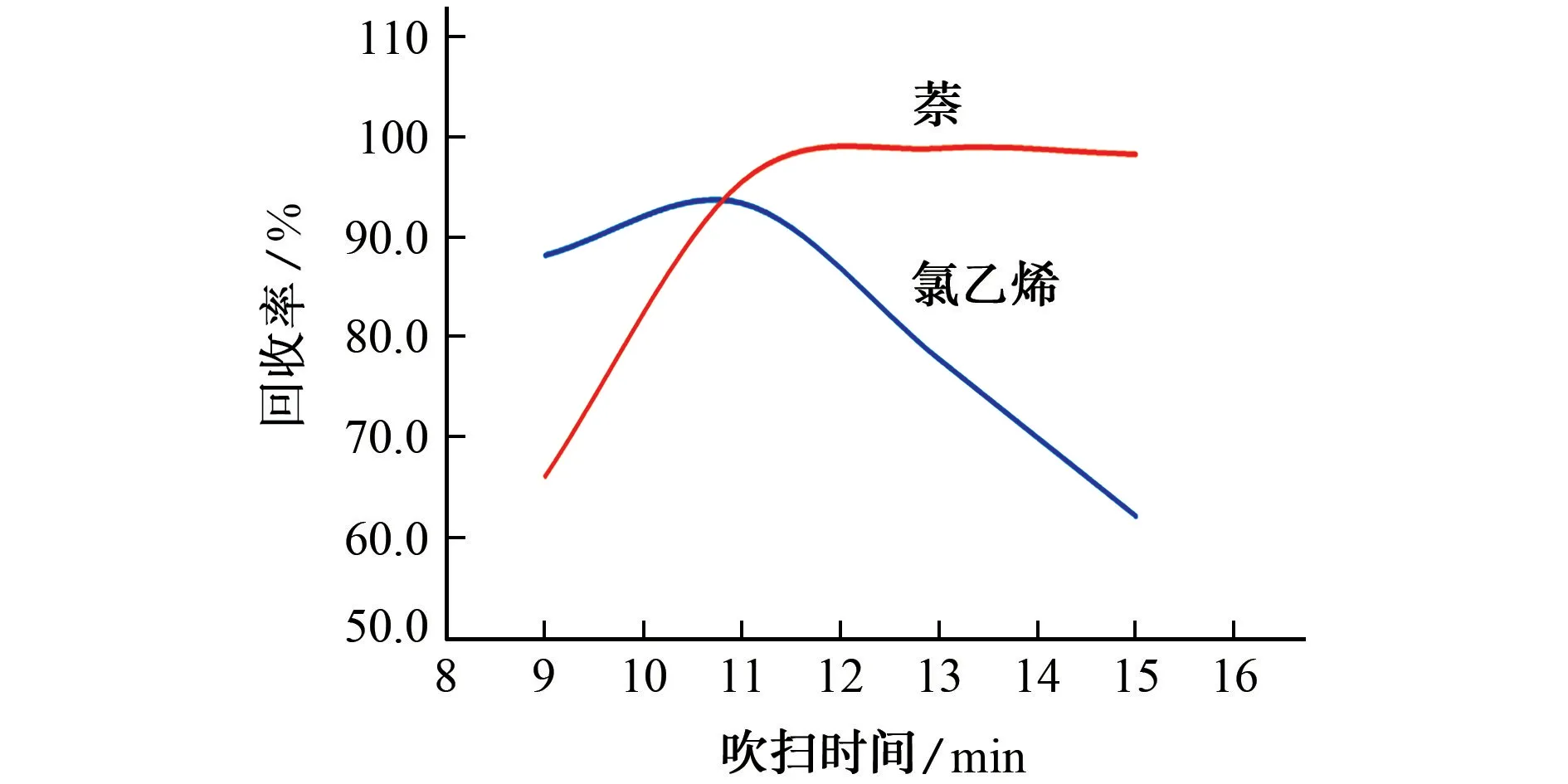
圖2 吹掃時間對氯乙烯、萘回收率的影響Fig.2 Effect of purging time on recovery of vinyl chloride and naphthalene
2.3 脫附溫度和脫附時間的優化
試驗采用Tekmar 9#新型捕集阱(14-9908-403),在查閱相關參考文獻基礎上,考察了脫附溫度(190[4-5,8],200[3,6,9],220[10],250 ℃)對各目標物回收率的影響。結果顯示:當脫附溫度小于250℃時,各目標物回收率普遍較低;隨著脫附溫度的上升,各目標物回收率顯著增加,但脫附溫度過高會縮短捕集阱的壽命。綜合考慮,試驗選擇脫附溫度為250 ℃。
在脫附溫度250℃條件下及查閱相關參考文獻基礎上,試驗考察了脫附時間(0.5[5,10],1.0,1.5,2.0[14],4.0 min[2,6])對各目標物回收率的影響。結果顯示:脫附時間為1.5,2.0 min時,37種VOCs回收率整體較好;但當脫附時間為1.5 min時,萘、六氯丁二烯等高沸點VOCs的回收率小于70%;當脫附時間為2.0 min時,氯乙烯等低沸點VOCs的回收率較差。
因此,進一步選擇脫附時間為1.6,1.7,1.8 min進行試驗。結果發現,當脫附時間為1.7 min時,各目標物的回收率均達到最高,且高、低沸點VOCs(萘、氯乙烯)的RRF的RSD 均最小。因此,試驗選擇脫附時間為1.7 min。
2.4 烘烤溫度和烘烤時間的優化
Tekmar 9#新型捕集阱的最高使用溫度為350 ℃,為了清除殘留目標物,烘烤溫度應高于脫附溫度250 ℃,因此不再考慮215[3],220[4],240 ℃[5]等溫度。綜合清除效果及吸附劑壽命,試驗選擇烘烤溫度為280 ℃。
在烘烤溫度280 ℃條件下,測定目標物質量濃度為40μg·L-1的水樣,考察烘烤時間為6,8[8],10 min[3-5,10]對目標物殘留的影響,同時測定空白樣品。結果表明:烘烤時間為8,10 min時,均可消除樣品之間的相互影響。根據節省時間及延長捕集阱使用壽命的原則,試驗選擇烘烤時間為8 min。
2.5 色譜行為
按照儀器工作條件測定,37種VOCs混合標準溶液的總離子流色譜圖見圖3。
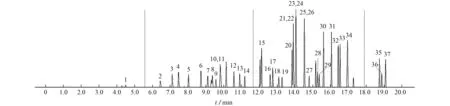
圖3 37種VOCs的總離子流色譜圖Fig.3 Total ion chromatogram of 37 VOCs
由圖3可知:20 min內可將37種VOCs完全分離,且目標物峰形較好。
2.6 校準曲線與檢出限
試驗分別采用校準曲線法和RRF 數據處理方法進行定量分析。地下水的線性范圍均為1~80μg·L-1,土壤樣品中目標物的線性范圍均為2~160μg·kg-1,線性回歸方程和相關系數的結果見表2。地下水、土壤樣品中目標物的RRF 及其RSD(n=8)見表2。
采用HJ 168-2010?環境監測 分析方法標準制修訂技術導則?中規定方法計算地下水、土壤樣品中目標物的檢出限(MDL=3.143sn,s為平行測定的標準偏差,n=7),結果見表2。峰號1~37對應的化合物名稱同表1。
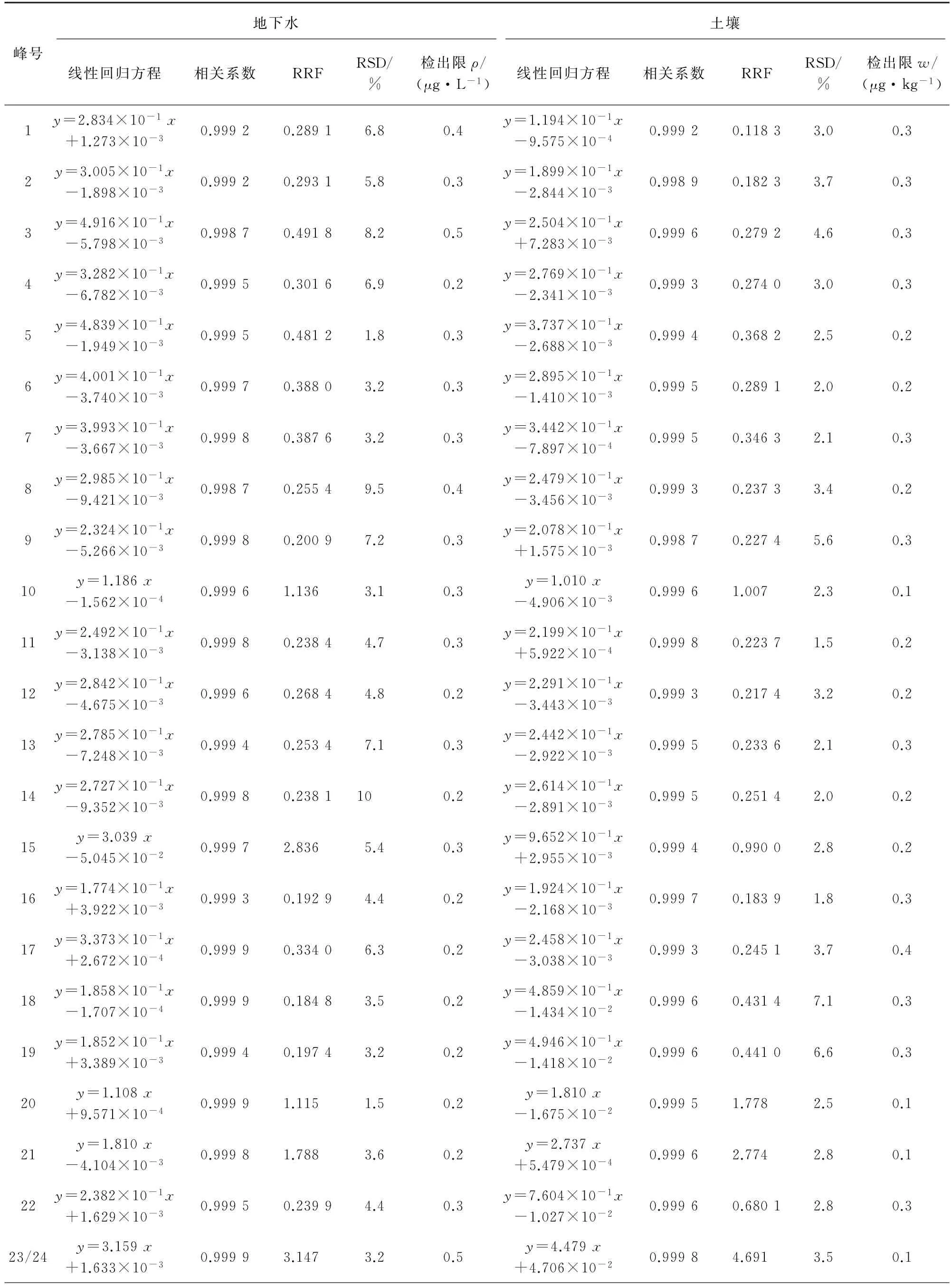
表2 地下水和土壤樣品中37種VOCs的兩種定量方法參數及檢出限Tab.2 Parameters of two quantitative methods and detection limits of 37 VOCs in groundwater and soil samples
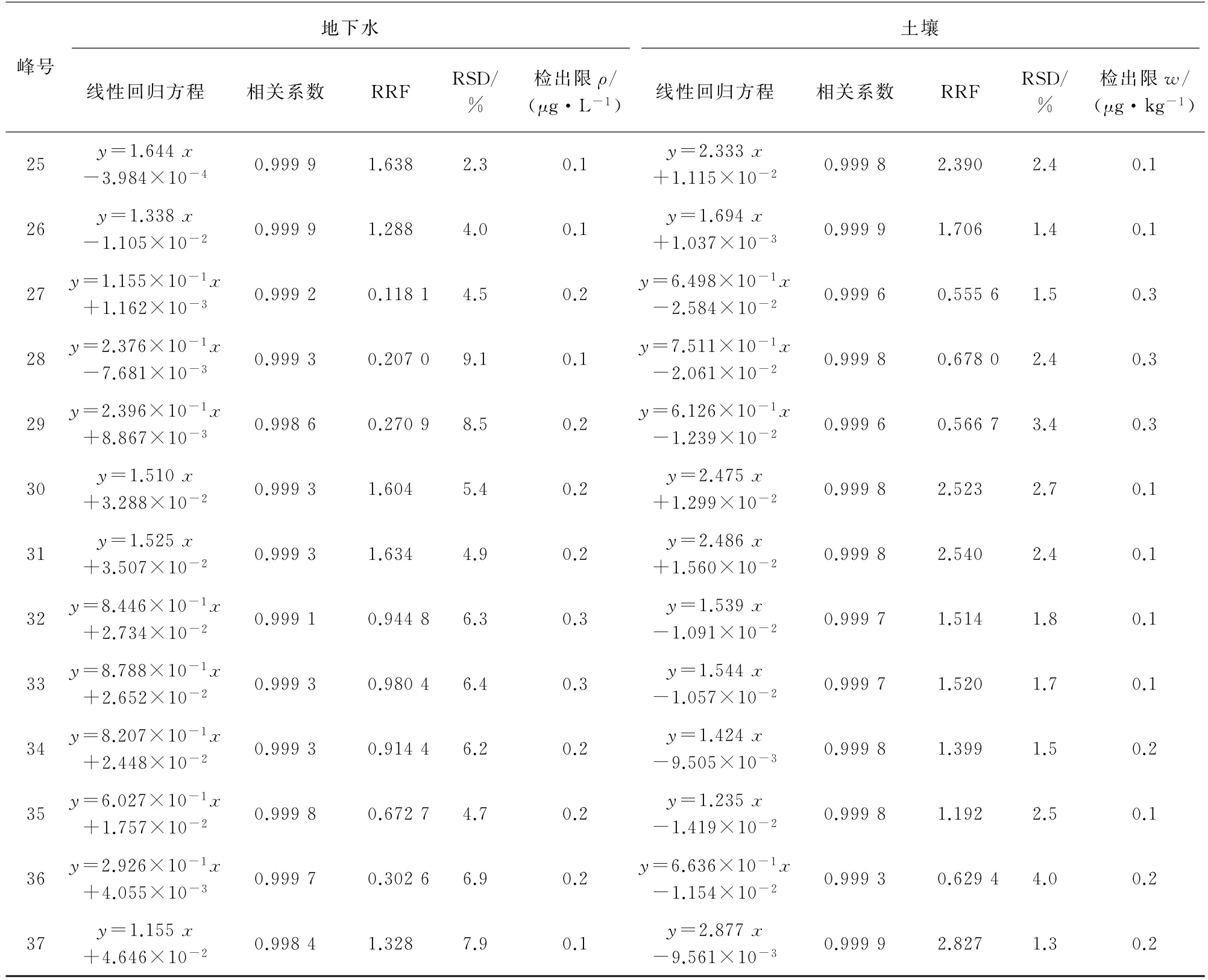
表2 (續)
由表2可知:地下水中各VOCs的檢出限均不大于0.5μg·L-1,滿足GB/T 14848-2017?地下水質量標準?對Ⅰ類水體的測定要求;土壤中各VOCs的檢出限均小于0.4μg·kg-1,遠低于GB 36600-2018?土壤環境質量建設用地土壤污染風險管理標準?中最低值0.05 mg·kg-1。兩種定量方法可分別滿足RRF的RSD 不大于10%及相關系數不小于0.998 0的要求,檢出限滿足相關標準的測定要求。在實際測定時可選擇校準曲線法或RRF數據處理方法,通用性均強。
2.7 精密度和回收試驗
以石英砂樣品為空白樣品,配制4,40,100μg·kg-1等低、中、高3個濃度水平的土壤基體加標樣品,同時對加標濃度水平為2,20,40μg·L-1的地下水樣進行測定,每個濃度水平平行測定7次,計算回收率和測定值的RSD,結果見表3。
由表3 可知:地下水樣中目標物的回收率為75.5%~112%,測定值的RSD 為0.85%~11%,其中低、中、高濃度水平的回收率依次為75.5%~108%,91.0%~109%,85.8%~112%;土壤基體加標樣品中目標物的回收率為82.0%~115%,測定值的RSD 為0.81%~7.9%,其中低、中、高濃度水平的回收率依次為83.5%~105%,82.0%~107%,82.2%~115%。回收率隨著加標濃度的增大而升高,其中二氯甲烷、四氯化碳的回收率偏低,原因可能是由于兩者沸點較低,在測定過程中易損失。
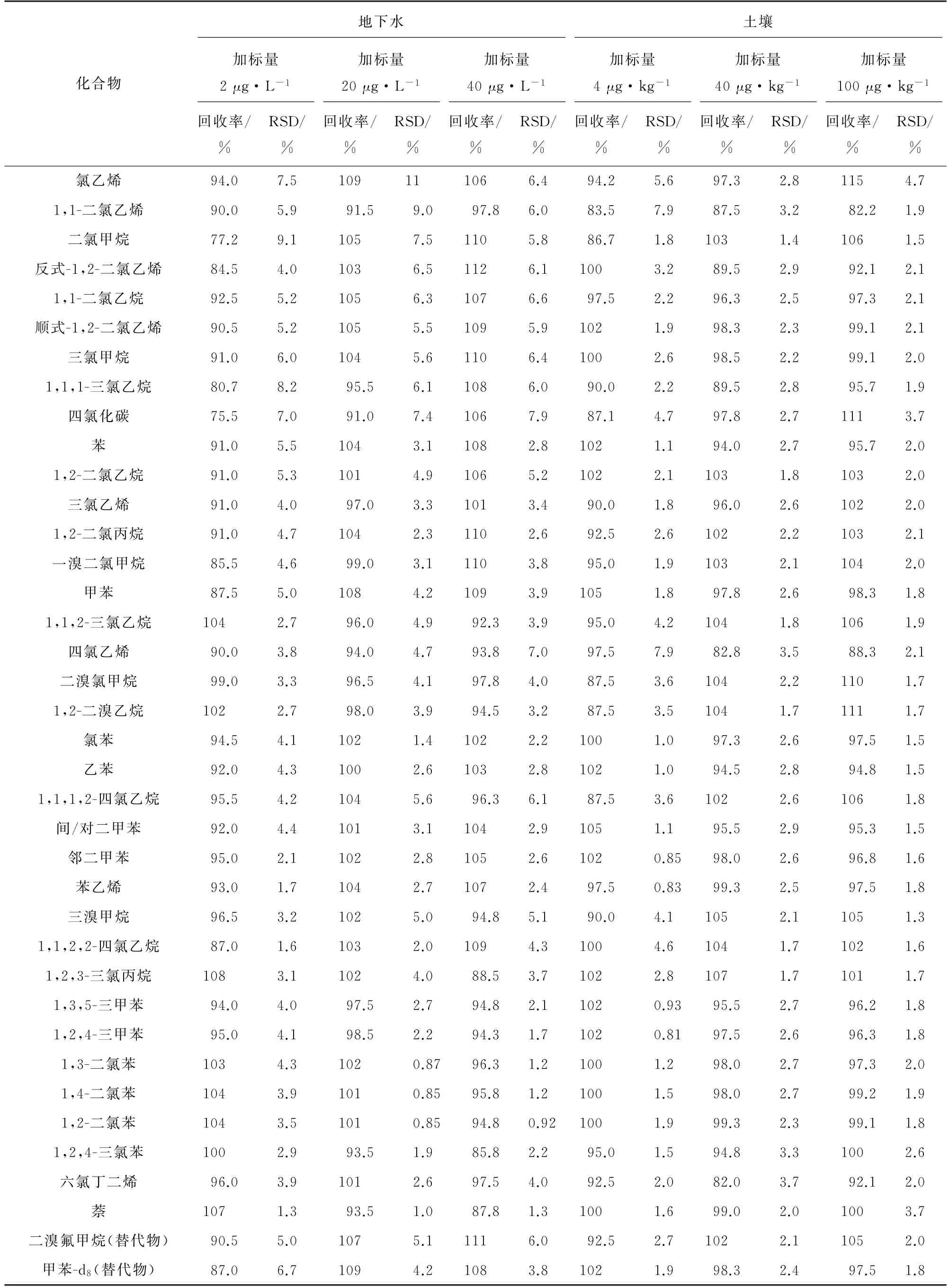
表3 精密度和回收試驗結果(n=7)Tab.3 Results of tests for precision and recovery(n=7)
2.8 樣品分析
按照試驗方法,對濟南市7個水源地(S1~S7)的土壤及地下水樣品進行分析。其中,僅在部分地下水樣品中檢出三氯甲烷、四氯化碳和四氯乙烯,結果見表4,而土壤樣品中的VOCs均未檢出。
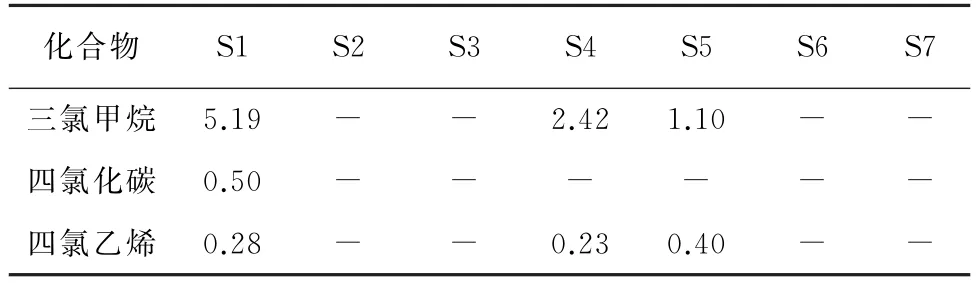
表4 樣品分析結果Tab.4 Analytical results of samples μg·L-1
本工作采用吹掃捕集-GC-MS快速測定同序列地下水和土壤中37種VOCs的含量,該方法穩定性好,操作簡便,在處理數據上具有普適性,適用于地下水和土壤單一介質或同時對VOCs進行批量檢測,可為地下水和土壤聯合修復提供技術支撐。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
