木犀草素分子印跡固相萃取柱的制備及應用
2021-08-26 10:42:10劉展眉張漢輝東方云肖裕澤程杏安蔣旭紅
食品研究與開發 2021年16期
劉展眉,張漢輝,東方云,肖裕澤,程杏安,蔣旭紅*
(1.廣州南洋理工職業學院教學科研部,廣東 廣州 510900;2.仲愷農業工程學院化學化工學院,廣東 廣州 510225)
黃酮類化合物是天然產物中富有活性的植物成分,在醫藥、食品等領域均有重要的意義。木犀草素是一種具有代表性的天然黃酮類化合物[1],具有抗氧化[2]、抗炎[3]、抗腫瘤[4]以及保護心血管[5]等多種生物活性。花生殼是木犀草素的最重要來源之一[6]。目前木犀草素的分離純化主要采用有機溶劑提取[7]、硅膠柱層析[8]或大孔吸附樹脂吸附[9]等,但由于植物中活性成分種類多樣,結構相似,所以存在著分離效果差、溶劑消耗量多以及操作繁瑣等缺點,利用傳統的分離純化技術得到高純度的目標分子存在較大難度[10-11]。因此研究和開發木犀草素的高效分離純化技術具有重要的意義。
分子印跡固相萃取技術是利用分子印跡聚合物作為固相萃取劑,選擇性分離目標物的技術[12]。相較于傳統的固相萃取,分子印跡固相萃取對目標化合物具有特異選擇性,能分離復雜樣品中的特定成分,并且具有較強的富集能力,檢測靈敏度高[13]。因此分子印跡-固相萃取現已成為分離純化的熱點之一,有著廣泛的應用前景[14-16]。高文姬[17]、肖淑娟等[18]采用本體聚合法制備木犀草素分子印跡聚合物,并以其為填料制備分子印跡固相萃取柱,從花生殼提取物中分離純化了木犀草素。本體聚合法制備的分子印跡聚合物存在模板分子除去困難、在研磨過程中容易破壞位點、印跡位點效率低等缺點,嚴重影響了分離柱效及選擇性[19],而采用沉淀聚合法制備分子印跡聚合物則不需研磨,顆粒大小較均勻,并且具有更多的均一識別位點[20-21]。
本研究以木犀草素為模板分子,α-甲基丙烯酸(α-methyl acrylic acid,MAA)為功能單體,乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA)為交聯劑,采用沉淀聚合法制備木犀草素分子印跡聚合物;并以其為固相萃取柱填料,制備分子印跡固相萃取柱,研究萃取柱對木犀草素的萃取性能,為花生殼中木犀草素的提純提供了一種高效的分離方法。
1 材料與方法
1.1 材料與試劑
木犀草素(純度97.81%):上海畢得醫藥科技有限公司;花生根莖:仲愷農業工程學院種子科學與工程研究所培育;α-甲基丙烯酸(MAA)、乙二醇二甲基丙烯酸酯(EGDMA)、偶氮二異丁腈(azobisisobutyronitrile,AIBN):阿拉丁試劑(上海)有限公司;所用試劑均為分析純。
1.2 儀器與設備
THZ-82水浴恒溫振蕩器:金壇市榮華儀器制造有限公司;HH-14數顯恒溫水浴鍋:常州澳華儀器有限公司;KH-45A電熱恒溫干燥箱:康恒儀器有限公司;ATY124電子天平:日本島津公司;Agilent-1200S高效液相色譜儀:美國安捷倫科技有限公司;Visiprep DL SPE真空固相萃取裝置:美國Supelco公司。
1.3 試驗方法
1.3.1 分子印跡聚合物的制備
以木犀草素為模板分子,MAA為功能單體,按1∶8質量比溶解于30 mL甲醇溶液中,加入6 mmol交聯劑EGDMA、100.0 mg引發劑AIBN到該混合溶劑中,超聲脫氣5 min,通氮氣5 min脫氧,密封后于60℃恒溫水浴中聚合24 h。所得固體以甲醇-乙酸溶液(8∶2,體積比)為溶劑進行索氏提取洗脫,直至洗脫液不含模板分子為止,再用甲醇反復洗滌聚合物除去殘留乙酸,真空干燥,得分子印跡聚合物(molecularly imprinted polymers,MIPs)。非分子印跡聚合物的制備除不加入木犀草素模板分子外,步驟與處理方法均與上述制備過程相同,得非分子印跡聚合物(non-imprinted polymers,NIPs)。
1.3.2 高效液相色譜條件(high performance liquid chroma-tography,HPLC)
色譜柱型號:CNW Athena C18-WP(4.6 mm×250 mm,5 μm);流動相:乙腈-水(30∶70,體積比),柱溫:28℃,流速:1.0 mL/min,紫外檢測波長:350 nm,進樣量:20 μL。
1.3.3 木犀草素標準曲線
配制1.0 mg/mL木犀草素標準儲備液,甲醇稀釋,制備 2、4、8、12、16、20、28、40 μg/mL 的木犀草素標準工作液,按照1.3.2高效液相色譜條件測定峰面積,繪制木犀草素的標準曲線。
1.3.4 木犀草素分子印跡聚合物的吸附性能
1.3.4.1 分子印跡聚合物的動態吸附試驗
準確稱取30.0 mg MIPs于20 mL具塞錐形瓶中,加入10 mL 1.0 mmol/L的木犀草素標準液,置于25℃恒溫水浴中振蕩,每隔一段時間(0.5、2、4、5、6、8 h),取出MIPs上清液,通過HPLC測定其中木犀草素的含量,繪制木犀草素吸附量Q(μmol/g)與吸附時間(h)之間的變化曲線。其中吸附量Q按下式(1)進行計算。

式中:Q為吸附量,μmol/g;Co為底物的初始濃度,μmol/mL;Ce為平衡后底物的濃度,μmol/mL;V 為吸附溶液的體積,mL;m為吸附劑印跡聚合物微球的質量,g。
1.3.4.2 分子印跡聚合物的選擇性考察
分別稱取30.0 mg MIPs與NIPs置于20 mL具塞錐形瓶中,加入10 mL 1.0 mmol/L的木犀草素標準液,置于25℃恒溫水浴中振蕩吸附8 h,取上清液,利用HPLC檢測木犀草素的含量,參照袁波的方法[22],印跡因子IF按下式(2)進行計算。

式中:IF為印跡因子;QMIPs為分子印跡聚合物對木犀草素的吸附量,μmol/g;QNIPs為非分子印跡聚合物對木犀草素的吸附量,μmol/g。
1.3.5 花生殼提取物的制備
稱取花生殼粉末8.0 g,加入160 mL乙醇-水溶液(8∶2,體積比),置于80℃恒溫水浴中萃取3 h,將得到的乙醇提取液旋蒸至近干得花生殼提取液,自然干燥得花生殼乙醇提取物,密封放入干燥器中備用。
1.3.6 分子印跡固相萃取柱的制備
在固相萃取柱(solid phase extraction,SPE)中通過干法分別裝入約0.3 g MIPs與NIPs填料,確保填料緊實平整,再加入少許石英砂于MIPs上方固定。裝柱完畢后,加入10 mL甲醇潤濕柱子。取2 mL花生殼提取液溶解于不同體積比的甲醇-水溶液后,裝入固相萃取柱,以不同體積比的甲醇-水溶液進行淋洗除去雜質,真空抽干,以25 mL不同種類的甲醇-酸溶液進行洗脫,收集淋洗液及洗脫液,HPLC檢測其中木犀草素的含量,木犀草素在固相萃取柱中的保留率A、洗脫率B 按下式(3)、(4)進行計算。
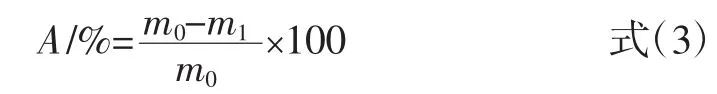
式中:A為木犀草素的保留率,%;m0為上樣液中木犀草素的質量,μg;m1為流出液中木犀草素的質量,μg。

式中:B為木犀草素洗脫率,%;m0為上樣液中木犀草素的質量,μg;m1為洗脫液中木犀草素的質量,μg。
2 結果與分析
2.1 木犀草素標準曲線
根據木犀草素的濃度C(μg/mL)與色譜圖峰面積A(mAU)之間的對應關系,建立線性方程,繪制相對的標準曲線:y=72.026x+19.419,相關系數R2=0.999 2。木犀草素標準曲線見圖1。
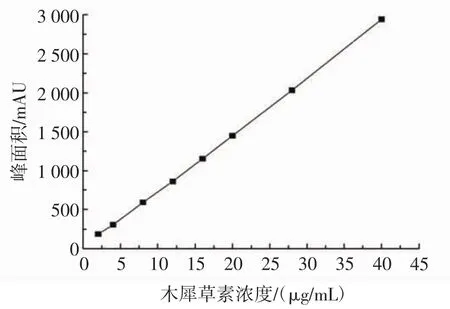
圖1 木犀草素標準曲線Fig.1 The standard curve of luteolin
2.2 分子印跡聚合物制備條件的優化
2.2.1 模板分子與功能單體的摩爾比選擇
以木犀草素為模板分子,α-甲基丙烯酸(MAA)為功能單體,固定交聯劑EGDMA用量為5 mmol/L,引發劑AIBN用量為100.0 mg,分別以不同摩爾比的木犀草素與 MAA(1∶4、1∶6、1∶8、1∶10、1∶12)制備 MIPs,考察模板分子與功能單體的摩爾比對MIPs吸附量的影響,結果見圖2。
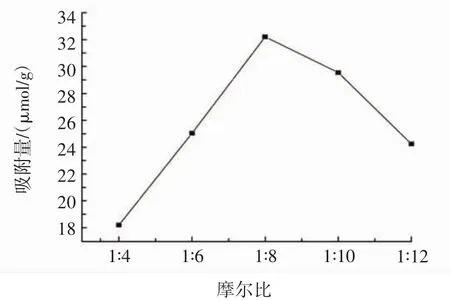
圖2 模板分子與功能單體的摩爾比對MIPs吸附量的影響Fig.2 Effect of the molar ratio of template to monomer on MIPs adsorption capacity
由圖2可知,隨著MAA用量的增加,MIPs的吸附量逐漸增加,當木犀草素與MAA的摩爾比為1∶8時,MIPs的吸附量達到最大值32.22 μmol/g,而隨著MAA用量的繼續增加,MIPs的吸附量逐漸減少。這是因為當功能單體MAA用量較少時,只有少量的木犀草素能與功能單體結合形成復合物,還有大量未結合的印跡分子存在,制備的MIPs中只有較少的木犀草素選擇性吸附位點,導致吸附量較少;而隨著MAA用量的增加,木犀草素與MAA間形成更多的選擇性吸附位點,吸附量逐漸增加,但MAA用量過多時,過量的MAA會導致非選擇性的結合位點增加,還會引發自身分子的聚合,導致MIPs中木犀草素的吸附位點減少,吸附量減少。因此,木犀草素與MAA的最佳摩爾比為1∶8。
2.2.2 交聯劑用量的選擇
固定木犀草素與MAA的摩爾比為1∶8,引發劑AIBN用量為100.0 mg,分別以不同用量的交聯劑EGDMA(2、4、6、8、10 mmol)制備 MIPs,考察交聯劑用量對MIPs吸附量的影響,結果見圖3。
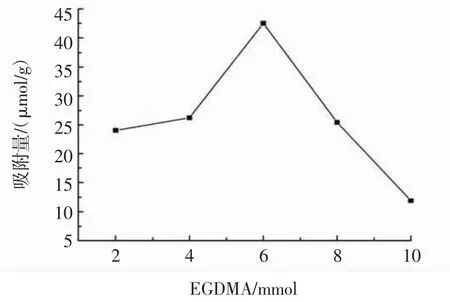
圖3 交聯劑用量對MIPs吸附量的影響Fig.3 Effect of the cross-linker dosage on MIPs adsorption capacity
由圖3可知,隨著交聯劑EGDMA用量的增加,MIPs的吸附量逐漸增加,當交聯劑EGDMA的用量為6 mmol時,MIPs的吸附量達到最大值42.56 μmol/g,而隨著EGDMA用量的繼續增加,MIPs的吸附量逐漸減少。這是因為當交聯劑EGDMA用量較少時,低交聯劑用量會使MIPs的印跡空穴剛性過低,洗脫后難以維持空穴原來的形狀和大小,導致木犀草素的識別效果不佳,吸附量較少;而隨著EGDMA用量的增加,MIPs的印跡空穴剛性增強,減少聚合物在不同溶劑中的溶脹,但當EGDMA的用量過多時,木犀草素在聚合物體系內的傳質會被高度交聯的網絡阻斷,導致分離效果下降,吸附量減少。因此,交聯劑EGDMA的最佳用量為6 mmol。
2.3 木犀草素分子印跡聚合物的吸附性能
2.3.1 分子印跡聚合物的動態吸附性能
用動態吸附試驗考察聚合物MIPs的吸附性能,繪制動態吸附曲線,結果如圖4。
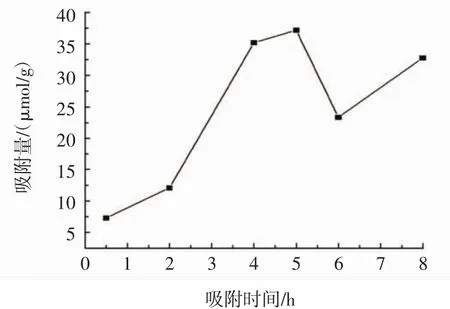
圖4 MIPs動態吸附曲線Fig.4 Dynamic adsorption curve of MIPs
由圖4可知,MIPs在前5 h內吸附量先迅速增大,后緩慢上升,在5 h時MIPs達到最大吸附量37.22 μmol/g,而5 h后,吸附量出現先下降后上升的現象。這是由于在吸附的初期,MIPs的結合位點未達到飽和狀態,吸附量隨著吸附時間的增加而迅速上升;當吸附量增大到一定程度時,溶液中木犀草素的濃度降低,MIPs出現外吐現象,使得吸附量降低,降低到一定程度時MIPs又重新開始吸附木犀草素。因此,MIPs在5 h時吸附量最大且基本達到吸附平衡。
2.3.2 分子印跡聚合物選擇性吸附性能
試驗結果表明,在1.0 mmol/L的木犀草素標準液中,MIPs的吸附量可達44.75 μmol/g,而NIPs的吸附量僅有 36.38 μmol/g,印跡因子按式(2)進行計算,得IF=1.23。MIPs比NIPs具有更強的選擇吸附性,這是由于NIPs在合成的過程中并沒有加入相應的模板分子,無法形成具有特異性吸附的空穴,所以NIPs的吸附主要是物理吸附作用,而合成的MIPs除了具有物理吸附外,還有特異性吸附,因此它的吸附量較NIPs高。
2.4 分子印跡-固相萃取條件的優化
2.4.1 上樣溶劑的選擇
由于木犀草素在甲醇溶液中溶解性較好,同時花生殼中木犀草素的提取以甲醇-水溶液為溶劑,因此試驗分別以不同體積比的甲醇-水溶液(0∶100、30∶70、40∶60、50∶50、60∶40、70∶30、100∶0)作為上樣溶劑,考察其對木犀草素保留率的影響,結果見圖5。
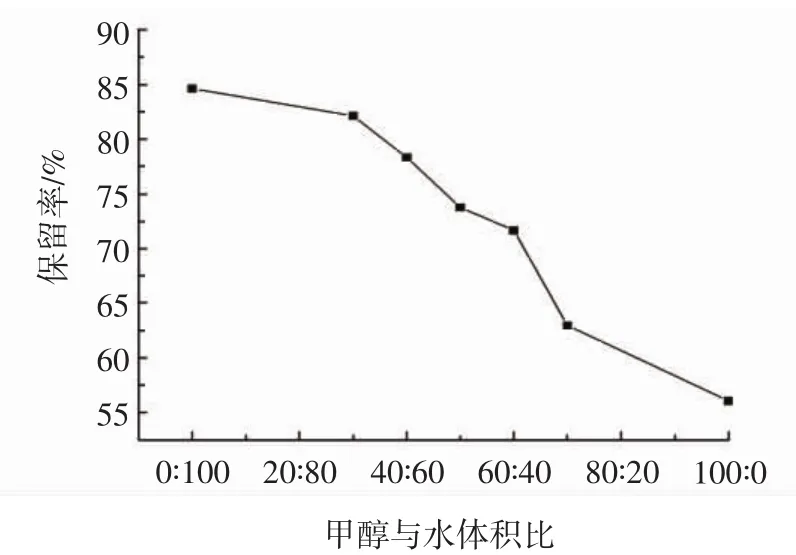
圖5 上樣溶劑對木犀草素保留率的影響Fig.5 Effect of sample solvent on luteolin retention rate
由圖5可知,隨著溶劑中甲醇含量的增加,木犀草素的保留率逐漸下降。當采用純水作為上樣溶劑時,木犀草素的保留率達到最高84.63%,而采用純甲醇作為上樣溶劑時,木犀草素的保留率達到最低56.06%。這是由于木犀草素在甲醇中溶解度較大,當上樣溶劑中甲醇濃度較高時,木犀草素容易隨著上樣液流出萃取柱,造成樣品的損失,保留率下降;而甲醇濃度過低時,則溶劑極性過大,容易影響聚合物中印跡空穴與木犀草素物質間的氫鍵作用[23];且甲醇濃度過低不利于粗提取物的溶解,造成上樣過程中部分提取物析出,進行淋洗時,容易隨著淋洗液流出,影響萃取效果。故選用甲醇-水溶液(50∶50,體積比)作為上樣溶劑。
2.4.2 淋洗液的選擇
試驗以10 mL不同體積比的甲醇-水溶液(10∶90、20∶80、30∶70、35∶65、40∶60、50∶50)作為淋洗液,考察其對木犀草素保留率的影響,結果見圖6。
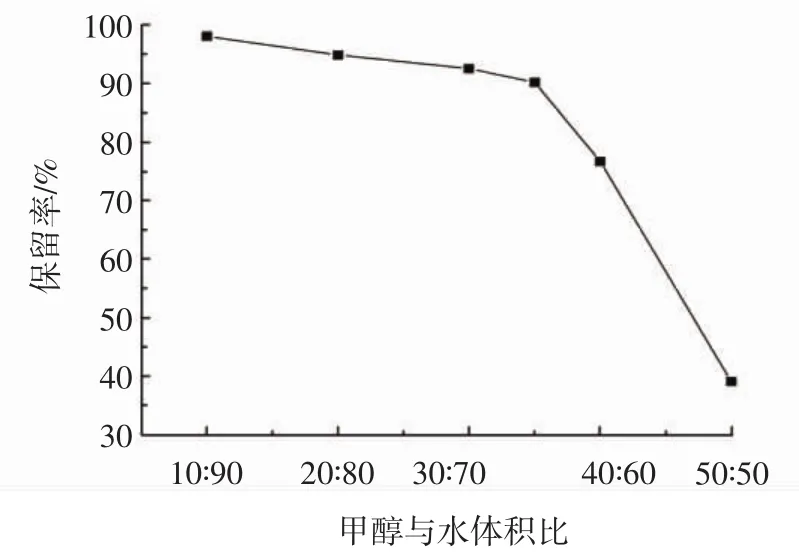
圖6 淋洗液對木犀草素保留率的影響Fig.6 Effect of abluent solvent on luteolin retention rate
由圖6可知,隨著甲醇含量的增加,木犀草素的保留率呈現下降的趨勢。當采用甲醇-水溶液(10∶90,體積比)作為淋洗液時,木犀草素的保留率達到最高98.11%,當采用甲醇-水溶液(50∶50,體積比)時,保留率達到最低39.08%。且當甲醇體積占比大于35%時,木犀草素的保留率快速下降,這是由于甲醇含量的增加,使木犀草素在淋洗液中的溶解度大幅上升,容易隨著雜質一起流出,造成保留率驟減。而甲醇-水體積比處于 10∶90~35∶65 時,木犀草素的保留率為 98.11%~90.24%,相對影響較小,因此選用甲醇-水溶液(35∶65,體積比)作為淋洗液。
2.4.3 淋洗液用量的選擇
試驗以 5、10、15、20、25 mL 的甲醇-水溶液(35∶65,體積比)作為淋洗液,考察其對木犀草素保留率的影響,結果見圖7。
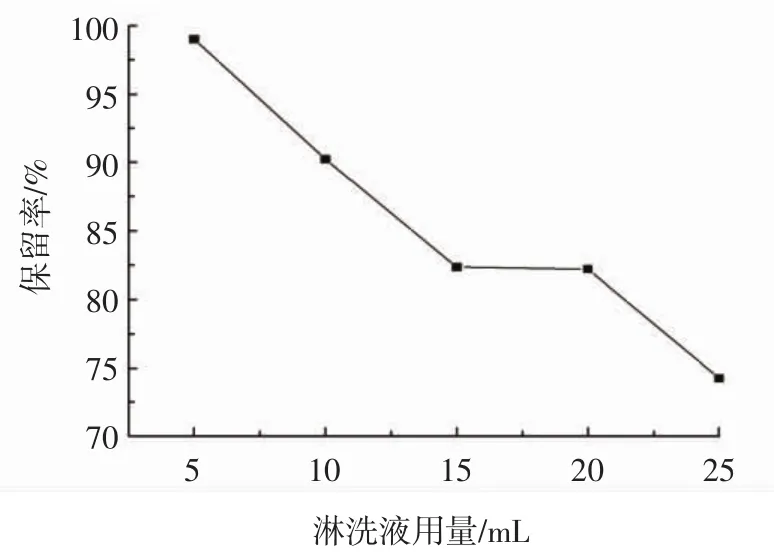
圖7 淋洗液用量對木犀草素保留率的影響Fig.7 Effect of dosage of abluent solvent on luteolin retention rate
由圖7可知,隨著淋洗液用量的增加,木犀草素的保留率呈現下降的趨勢。當淋洗液用量為5 mL時,木犀草素保留率達到最高99.00%,而當淋洗液用量為25 mL時,保留率達到最低74.25%。淋洗液用量在5 mL~15 mL時,大部分雜質以及少量木犀草素隨淋洗液流出,而當淋洗液用量為15 mL~25 mL時,大量木犀草素開始溶解于淋洗液中,隨著淋洗液流出,造成保留率的下降。但是淋洗液用量過低易造成雜質殘留。因此選用15 mL的甲醇-水溶液(35∶65,體積比)作為最佳淋洗液。
2.4.4 洗脫液的優化
試驗分別選取25 mL甲醇-乙酸溶液(80∶20,體積比)、甲醇-甲酸溶液(80∶20,體積比)、甲醇-乙酸溶液(90∶10,體積比)、純甲醇溶液作為洗脫液,考察其對木犀草素洗脫率的影響,結果見圖8。
由圖8可知,當采用純甲醇作為洗脫液時,木犀草素的洗脫率達到最低67.76%,當采用體積比80∶20的甲醇-甲酸溶液作為洗脫液時,洗脫率達到最高85.57%。這是由于MIPs主要通過氫鍵作用吸附木犀草素,在保持相同洗脫劑用量的前提下,加入少量的有機酸易于破壞木犀草素與MIPs間的氫鍵作用,洗脫率上升。甲酸的酸性比乙酸強,所以80%甲醇-甲酸洗脫率最高,因此選用25 mL的體積比80∶20的甲醇-甲酸溶液作為最佳洗脫液。
2.5 花生殼提取物中木犀草素分子印跡-固相萃取純化
按照1.3.2的HPLC條件對花生殼提取物上樣液、固相萃取淋洗液以及洗脫液進行檢測,木犀草素標準品、花生殼提取物上樣液、固相萃取淋洗液以及洗脫液的HPLC色譜圖見圖9。液相色譜的分析結果見表1。
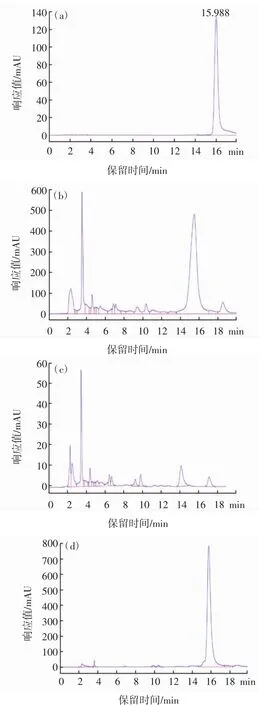
圖9 HPLC色譜圖Fig.9 The HPLC figure
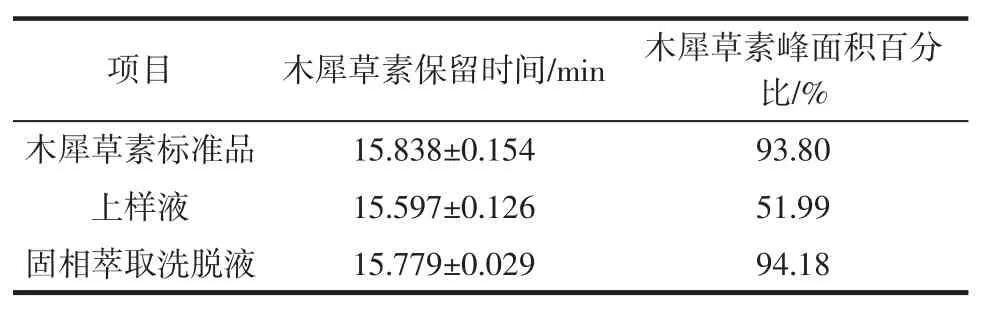
表1 液相色譜的分析結果Table 1 Analysis results of HPLC
由圖9、表1可知,木犀草素標準品的平均保留時間是(15.838±0.154)min;花生殼提取物上樣液中木犀草素的平均保留時間是(15.597±0.126)min,除此之外還存在大量雜質峰;固相萃取洗脫液中木犀草素的平均保留時間是(15.779±0.029)min,雜峰顯著減少。固相萃取淋洗液在保留時間15 min~16 min無木犀草素的特征峰出現,只存在大量雜峰,說明固相萃取的淋洗液可較好地除去雜質。花生殼提取物上樣液中木犀草素的峰面積占總面積51.99%,經分子印跡-固相萃取分離純化后,木犀草素的峰面積占總面積94.18%,提高了42.19%。
3 結論
采用沉淀聚合法制備了木犀草素分子印跡聚合物,該印跡聚合物對木犀草素具有優良的吸附效果以及選擇性。以其為填料制備了木犀草素分子印跡固相萃取柱,確定了固相萃取柱的最優上樣溶劑為甲醇-水溶液(50∶50,體積比)、淋洗液為甲醇-水溶液(35∶65,體積比)、洗脫液為體積比80∶20的甲醇-甲酸溶液,用此固相萃取柱對花生殼提取物中的木犀草素進行分離純化,該固相萃取柱對木犀草素表現出較好的選擇性吸附效果,可分離純化出相對純度為94.18%的木犀草素,為花生殼中木犀草素的分離純化提供了一種高效的分離方法。
