小腸系膜巨大叢狀神經纖維瘤1例
2021-08-19 02:29:10陳妹瓊李淑英
安徽醫學 2021年7期
陳妹瓊 李淑英
1 病例資料
患者,男性,15歲,因“間斷腹痛1個月”入院。患者自2017年9月15日無明顯誘因出現腹痛,以右側腹部為重,呈持續性絞痛,伴惡心嘔吐。外院全腹CT平掃+增強示:沿腸系膜及乙狀結腸系膜走行區不規則囊性低密度影,其內夾雜較多血管影,考慮良性脈管源性占位,血管淋巴管瘤可能。予對癥支持治療后腹痛減輕。于2017年10月17日在本院行手術治療,術中探查結腸及系膜未見腫物,中段小腸系膜可見一大小約30 cm×20 cm×3 cm餅狀多發結節融合腫物,無浸潤,局部腸壁水腫。遂行小腸系膜腫物切除術。切除標本病理檢查:送檢小腸腸管長60 cm,腸管直徑1.5~2 cm,小腸系膜處見一腫塊(見圖1),腫塊大小24 cm×18 cm×4 cm,表面可見包膜,切面見腫塊由大量扭曲、迂回的實性管狀結構組成,色灰白,膠凍狀,質中;低倍鏡下見腫塊位于小腸系膜,未累及小腸漿膜面,由迂曲、膨大的神經束組成,呈叢狀結構(見圖2);高倍鏡下見瘤細胞呈梭形,核呈尖細波浪狀,間質黏液樣變性(見圖3)。免疫組化:腫瘤細胞Vimentin、CD99、 S-100(見圖4)均陽性,EMA陽性的細胞分布于叢狀結構的邊緣, Ki-67增殖指數<1%。病理診斷 :(小腸系膜)叢狀神經纖維瘤。患者術后定期復查腹盆CT,術后3年未見復發。
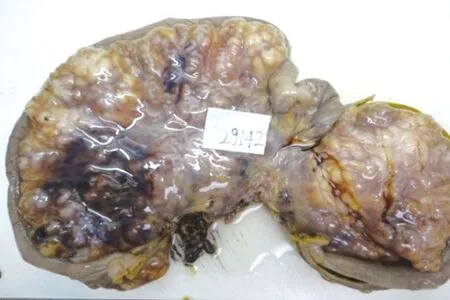
圖1 腫塊大體表現注:腫塊位于小腸系膜
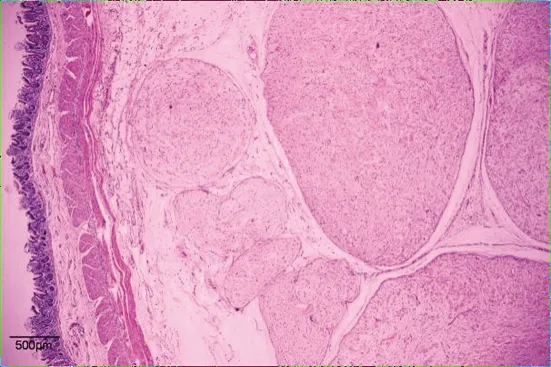
圖2 腫塊組織病理切片(HE染色)注:腫塊由大小不等的叢狀結構組成
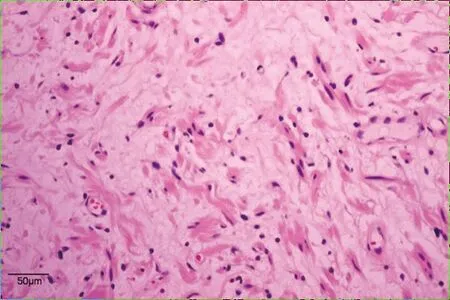
圖3 腫塊組織病理切片(HE染色 )注:瘤細胞呈梭形,核尖細,間質黏液樣變性

圖4 腫塊組織病理切片(IHC染色) 注:腫瘤細胞核及胞漿呈棕黃染色,S-100陽性表達
2 討論
叢狀神經纖維瘤(plexiform neurofibroma, PNF)在臨床比較少見,是神經纖維瘤的一個亞型,常發生于兒童,好發于頭頸部,引起畸形,也可見于軀干、四肢,少數發生于胃、膽管、胰腺、肝、膀胱等處。發生于腸系膜的PNF極少見,查閱國內外文獻,僅見3例報道,3例患者均為男性,年齡11~20歲,其中2例發生于小腸系膜,另一例累及腸系膜及直腸,患者均行手術切除,其中累及直腸者因病變廣泛,未完全切除。本病幾乎均伴發于Ⅰ型神經纖維瘤病(neurofibromatosis-1, NF1),并有惡變的傾向。本例患者胸壁、腹部及雙腿部可見十余處大小不等的圓形或橢圓形咖啡色斑塊,直徑1.5~4 cm,達到NF1的診斷標準,但患者無家族遺傳史及無皮膚神經纖維瘤病史。文獻報道,近一半的NF1患者最終發展成PNF,影響周圍大神經,PNF通常會在兒童、青少年和成年期持續發展,導致終身畸形、殘疾和死亡。
PNF的確診主要依靠病理學檢查及臨床表現。PNF具有顯著的叢狀結構和明顯的間質黏液樣變性,需與叢狀神經鞘瘤、神經鞘黏液瘤、創傷性神經瘤、叢狀纖維組織細胞瘤區別診斷,根據鏡下形態、免疫組化及臨床表現均可鑒別。腸系膜PNF罕見,易導致術前誤診。PNF多伴有皮膚表現,如皮膚結節、牛奶咖啡斑,或有虹膜錯構瘤、骨損害等,結合患者的臨床表現及影像學特征,要考慮到該病的可能。
PNF的治療以手術切除為主。然而,腫瘤常與神經、血管或其他臟器解剖結構關系密切,導致無法手術切除或不能完全切除。雖然目前還沒有可治愈PNF的藥物,但是一項臨床一期研究報道,使用MEK抑制劑治療后,腫瘤明顯縮小;另有一項二期臨床研究認為,MEK抑制劑曲美替尼可以納入PNF的標準治療。因此,MEK抑制劑可能在叢狀神經纖維瘤的治療中發揮重要作用。
