貴金屬摻雜銳鈦礦TiO2光催化性能的第一性原理研究
2021-08-16 07:27:12吳方棣楊自濤胡家朋李素瓊
原子與分子物理學(xué)報(bào) 2021年3期
關(guān)鍵詞:體系
吳方棣,楊自濤,胡家朋,李素瓊
(1.武夷學(xué)院福建省生態(tài)產(chǎn)業(yè)綠色技術(shù)重點(diǎn)實(shí)驗(yàn)室,武夷山 354300;2.嘉應(yīng)學(xué)院生命科學(xué)學(xué)院,梅州 514015)
1 引 言
TiO2作為最早開發(fā)利用的光催化材料,由于其廉價(jià)、無毒、化學(xué)穩(wěn)定性好等優(yōu)點(diǎn),一直是光催化領(lǐng)域的首選.但由于其能帶寬度較大(銳鈦礦TiO2為3.2 eV),只能利用紫外光和對可見光的利用率低等缺陷,從而使其應(yīng)用的成本較高,極大限制其在光催化過程的應(yīng)用.因此減小能帶寬度提高其對可見光的利用率一直是國內(nèi)外學(xué)者研究的熱點(diǎn).
對TiO2進(jìn)行摻雜改性是減小能帶寬度,提高其可見光利用率的一個(gè)有效手段,在摻雜改性中,又以金屬及其復(fù)合物的形式最為常見.李沙沙等[1],采用水熱法制備了La、Co共摻雜的金紅石相TiO2復(fù)合光催化劑,XRD測試表面摻雜沒有改變TiO2的的晶型結(jié)構(gòu),仍為金紅石相,但提高了TiO2表面缺陷濃度,有效的提高了材料的光催化性能.劉月等[2]對Y,La,Gd等16種稀土摻雜銳鈦礦型TiO2進(jìn)行了幾何結(jié)構(gòu)、摻雜能帶、電子態(tài)密度等的理論研究,結(jié)果表明:Y,La,Gd,Lu,Ce,Eu,Yb和Tb摻雜有利于提高TiO2的光催化性能.Altomare[3]通過正十二烷基三甲基氯化銨預(yù)制穩(wěn)定的金屬(Pt、Pd、Au和Ag)納米顆粒膠態(tài)懸浮液,讓其在P25 TiO2分散液中沉積,制備得到不同金屬摻雜的TiO2光催化劑.研究表明貴金屬納米顆粒的摻雜相當(dāng)于電子陷阱,確保更好地分離光生電子對,從而提高空穴介導(dǎo)的氧化反應(yīng)速率,有利于光催化氧化過程.Luo等人[4]采用溶膠-凝膠法制備了La/Fe/TiO2復(fù)合光催化劑.其制備的TiO2沒有摻雜時(shí)為金紅石相,摻雜La/Fe時(shí)為銳鈦礦.La/Fe/TiO2復(fù)合光催化劑比純的TiO2有更強(qiáng)的可見光響應(yīng)能力、更大的比表面積和更規(guī)整的形貌;其對氨氮廢水也有更高的催化活性.Dozzi[5]等采用兩種不同的沉積方法,制備了Pt/TiO2,Ru/TiO2及Pt-Ru/TiO2光催化劑.熒光分析結(jié)果表明,Pt的摻雜可以讓TiO2吸收光譜紅移,提高可見光的利用率.HRTEM分析結(jié)果表明,不同的沉積路徑,Pt在TiO2上的分布不同.Huang等[6]進(jìn)行了La、N共摻雜銳鈦礦TiO2體系的理論研究,分析了摻雜增強(qiáng)光催化活性的機(jī)理,確定了提高光催化性能的摻雜方法.Khan[7]采用溶膠-凝膠法制備了兩種不同鐵濃度摻鐵TiO2納米顆粒,結(jié)果表明與純TiO2納米粒子相比,鐵摻雜TiO2納米粒子光學(xué)帶隙隨鐵含量的增加而紅移,在可見光照射下對亞甲基藍(lán)染料的光催化降解性能有所提高.Liu等[8]采用爆燃法一步合成有較高光催化性能的Ag修飾Ti3+摻雜TiO2納米片,Ag修飾的Ti3+摻雜TiO2復(fù)合材料具有豐富的超小Ag納米粒子和Ti3+離子,由于Ag納米粒子的等離子體效應(yīng),提高了光吸收能力和光生載流子的分離率,有效提高材料光催化降解有機(jī)污染物的能力.
在金屬摻雜改性中,貴金屬摻雜的實(shí)驗(yàn)研究報(bào)道較多,對其理論分析計(jì)算的報(bào)道還較少,本文以第一性原理對Ru、Pd、Pt、Ag和Au等貴金屬摻雜銳鈦礦TiO2進(jìn)行計(jì)算,計(jì)算摻雜前后的晶格參數(shù)、能帶結(jié)構(gòu)、態(tài)密度和光學(xué)性質(zhì)變化,從理論上分析貴金屬摻雜后對TiO2光催化性能的影響.
2 理論計(jì)算方法及模型
理論計(jì)算采用基于密度泛函理論的第一性原理方法,在Materials Studio 7.0中由CASTEP模塊完成[9].計(jì)算過程中選用廣義梯度近似(GGA)的超軟贗勢和PW91交換關(guān)聯(lián)函數(shù)[10].在倒格矢空間,經(jīng)收斂性測試后,Monkhorst-Pack的K點(diǎn)網(wǎng)格取為4×8×3,平面波截?cái)嗄?80 eV,自洽精度為2.0×10-6eV/atom.TiO2及其摻雜模型在性質(zhì)計(jì)算前進(jìn)行了幾何結(jié)構(gòu)的優(yōu)化.
研究中采用銳鈦礦相,空間點(diǎn)群為I41/amd的TiO2為原胞模型,構(gòu)建了2×1×1的純TiO2(Ti8O16)和貴金屬摻雜TiO2(Ti7MO16,其中M分別為Ru、Pd、Pt、Ag和Au)的超晶胞模型.貴金屬摻雜時(shí)分別用選定的貴金屬替代超胞中的一個(gè)Ti原子,摻雜替代位置如圖1所示,分別計(jì)算摻雜后晶體模型能帶結(jié)構(gòu)、態(tài)密度和光學(xué)性質(zhì).本次計(jì)算采用的價(jià)電子組態(tài)分別為:O:1s22s22p4、Ti:[Ar]3d24s2、Ru:[Kr]4d75s1、Pd:[Kr]4d10、Ag:[Kr]4d105s1、Pt:[Xe]4f145d96s1和Au:[Xe]4f145d106s1.

圖1 貴金屬摻雜銳鈦礦TiO2(Ti7 MO16)的晶體模型:M分別為Ru、Pd、Pt、Ag和AuFig.1 Crystal model of noble metal doped anatase TiO2(Ti7 MO16):M is Ru,Pd,Pt,Ag and Au respectively
3 結(jié)果分析與討論
3.1 摻雜前后的晶體結(jié)構(gòu)
為了檢驗(yàn)計(jì)算方法的準(zhǔn)確性,本文計(jì)算了銳鈦礦TiO2的晶格參數(shù)、體積模量和形成能,并與實(shí)驗(yàn)值進(jìn)行了比較,如表1所示.從表中可以看出,TiO2晶格參數(shù)的結(jié)果與實(shí)驗(yàn)值基本一致;形成能計(jì)算值為-9.91 eV,與實(shí)驗(yàn)值-9.78 eV誤差為1.3 %,體積模量計(jì)算值與實(shí)驗(yàn)值誤差為3.9 %,總體誤差均較小,說明本次采用的計(jì)算方法可行.
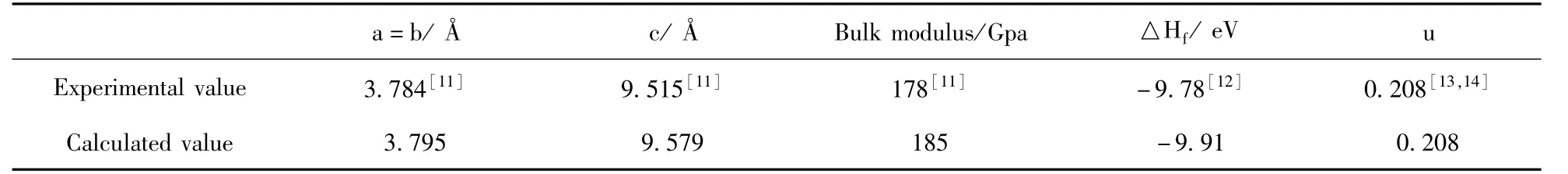
表1 銳鈦礦TiO2晶格參數(shù)Table 1 Lattice parameters of anatase TiO2
計(jì)算比較了摻雜貴金屬后的TiO2的晶格參數(shù)和體積變化,如表2所示.從表中可以看出,隨著貴金屬的摻雜,銳鈦礦TiO2的晶格體積都有不同程度的增大.這主要是由于摻雜TiO2后體系貴金屬與氧的鍵長發(fā)生了比較大的變化[15].計(jì)算得到的單純銳鈦礦TiO2的Ti-O徑向和軸向的鍵長分別為1.939?和1.995?,與實(shí)驗(yàn)值1.93?和1.98?基本一致[13,14].從表2可以看出,相比于單純銳鈦礦TiO2的Ti-O鍵長,不管是徑向還是軸向,摻雜貴金屬M(fèi)與氧的鍵長都要比其來的更大,從而導(dǎo)致了摻雜后晶格體積的增大.并且M-O鍵長越長,晶格體積越大.M-O鍵長大于Ti-O鍵長,也說明了摻雜的貴金屬M(fèi)-O的鍵能要小于Ti-O的鍵能,鍵能大小的變化也導(dǎo)致了晶格畸變的發(fā)生,這在一定程度上有利于提高材料的光催化性能.同時(shí),我們注意到,在摻雜后晶格體積增大,但也有某一軸向反而收縮的情況,如Pt、Pd摻雜后的c軸,Ru摻雜后的b軸,以及Ag摻雜后的a軸都出現(xiàn)了局部收縮的情況,其可能原因是不同貴金屬與TiO2之間的局部雜化[16]和原子半徑差異等綜合作用的結(jié)果.
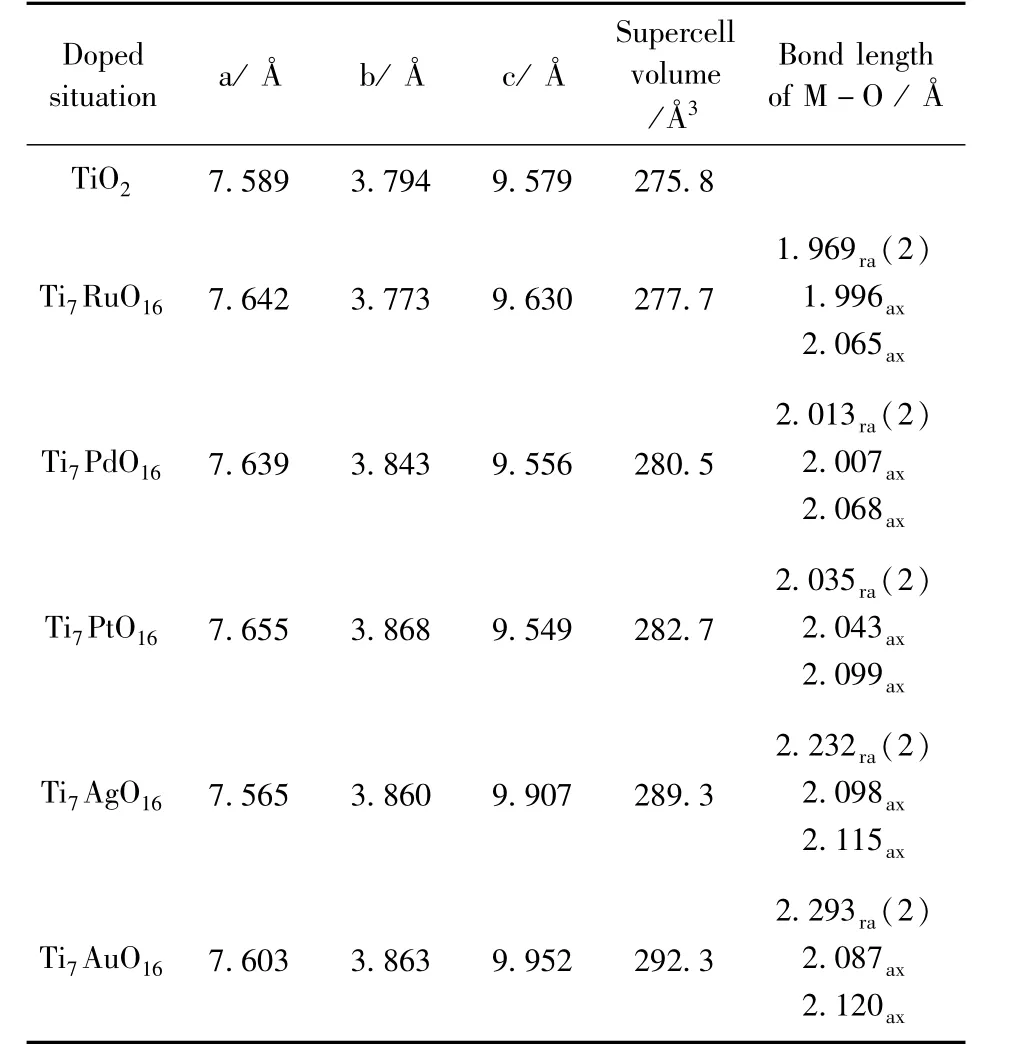
表2 摻雜前后TiO2的晶格參數(shù)和鍵長Table 2 Lattice parameters and bond lengths of undoped and noble metal doped anatase TiO2
為了進(jìn)一步分析摻雜貴金屬對晶格結(jié)構(gòu)的影響,本文計(jì)算了摻雜貴金屬原子和相連接氧原子的密立根電荷分布,結(jié)果如表3所示.對于單純銳鈦礦TiO2,其Ti和O的密立根電荷分布為1.33和-0.66,與Yu[15]等人的計(jì)算結(jié)果一致.從表3可以看出,5種摻雜貴金屬原子的電荷布居均小于單純銳鈦礦TiO2中Ti的1.33;相對應(yīng)的,與貴金屬原子相連接的氧原子的電荷布居均低于(絕對值)單純銳鈦礦TiO2中O的-0.66,說明摻雜的5種貴金屬原子的電負(fù)性要大于Ti原子的電負(fù)性,這有利于電荷在晶格內(nèi)的轉(zhuǎn)移和電子空穴的形成,對光催化過程是有利的.摻雜原子與Ti原子電負(fù)性的差異也進(jìn)一步導(dǎo)致了晶格畸變的產(chǎn)生.

表3 摻雜貴金屬原子和相連接氧原子的密立根電荷分布Table 3 Millikan charge distributions of doped noble metal atoms and connected oxygen atoms
3.2 摻雜的形成能
為了比較不同摻雜模型及不同條件下?lián)诫s體系的穩(wěn)定性,我們計(jì)算了摻雜體系在富鈦(Tirich)和富氧(O-rich)條件下的形成能.摻雜的形成能假定如(1)式反應(yīng)進(jìn)行[11]:

式中M為摻雜金屬(本文中為Ru、Pd、Pt、Ag和Au)形成能,形成能計(jì)算式如(2)式所示:

式中μ為對應(yīng)組分的化學(xué)勢.分別考慮了富鈦(Ti-rich)和富氧(O-rich)條件下的化學(xué)勢.
Ti-rich條件各元素化學(xué)勢計(jì)算如(3)式所示:
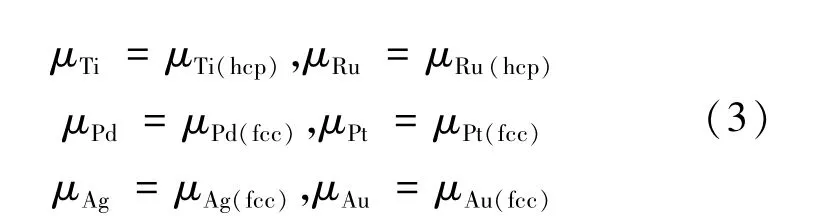
O-rich條件下各元素化學(xué)勢計(jì)算如(4)~(9)式所示:


計(jì)算結(jié)果如表4所示.從表中可以看出,在O-rich狀態(tài)下,Pd、Pt、Ag、Au(都屬于面心立方晶系,face-centered cubic)的摻雜更容易實(shí)現(xiàn),這與馬新國[17]等人研究的Pt和Au在銳鈦礦TiO2(101)面的空位吸附結(jié)果一致;而對于金屬Ru,其在Ti-rich條件下的摻雜更易實(shí)現(xiàn),可能原因是其與Ti一樣都是屬于密排六方晶體(hexagonal close packed lattice)的結(jié)構(gòu).從數(shù)值來看,在Orich條件下,摻雜形成能其均為負(fù)值,表明這些貴金屬摻雜TiO2是能夠?qū)崿F(xiàn)的,并且Ag、Au的形成能相較于其他貴金屬要小很多,說明其摻雜的熱力學(xué)穩(wěn)定性比其他貴金屬要好,與Pan[16]等人的研究結(jié)果一致.

表4 貴金屬摻雜TiO2形成能Table 4 Formation energies(in eV)of noble metal doped TiO2
3.3 銳鈦礦TiO2摻雜前后能帶結(jié)構(gòu)
銳鈦礦TiO2摻雜貴金屬前后能帶結(jié)構(gòu)如圖2所示.圖2(a)為純銳鈦礦TiO2能帶結(jié)構(gòu)圖,從圖中可以看出,其禁帶寬度為2.123 eV,與文獻(xiàn)[15,18]一致,小于實(shí)驗(yàn)值3.2 eV[19],這主要是由于GGA廣義梯度近似方法的誤差引起的[20-22],但該方法是有效可行的,計(jì)算結(jié)果的相對值是準(zhǔn)確的,禁帶寬度偏低不影響對能帶圖和態(tài)密度的分析.從圖2中可以看出,貴金屬摻雜后TiO2的能帶圖都發(fā)生了明顯的變化,Pd、Pt摻雜后的能帶間隙分別為0.812 eV和1.251 eV,與純銳鈦礦TiO2相比明顯減小,包秀麗[22]計(jì)算的Pt摻雜TiO2后的能帶間隙為1.32 eV,與本次計(jì)算結(jié)果接近,都明顯小于純TiO2的能帶間隙.從圖2(c)、2(d)可以看出,Pd、Pt摻雜后,能帶數(shù)明顯增多,導(dǎo)帶出現(xiàn)了明顯的下移,這有利于促進(jìn)價(jià)帶和導(dǎo)帶之間電子的躍遷,有利于價(jià)帶空穴的生成,對促進(jìn)TiO2的光催化氧化過程是有利的.從圖2(b)、2(e)和2(f)可以看出,Ru、Ag、Au摻雜TiO2后的帶隙消失,使材料表現(xiàn)出了一定的金屬屬性[16].根據(jù)半導(dǎo)體的摻雜理論,當(dāng)摻雜濃度較高時(shí),可使孤立的雜質(zhì)擴(kuò)展成為能帶,形成雜質(zhì)能帶[23].圖2(b)、2(e)和2(f)中,Ru、Ag和Au摻雜分別在禁帶中產(chǎn)生了3條、2條和2條的雜質(zhì)能級,Pd和Pt摻雜后同樣也在價(jià)帶頂和導(dǎo)帶底產(chǎn)生了明顯的雜質(zhì)能級.雜質(zhì)能級可在價(jià)帶電子躍遷中起過渡作用,減小電子躍遷所需的激發(fā)能量,從而拓寬了貴金屬摻雜后TiO2的光響應(yīng)波長范圍[23],這在后面的光譜分析中可得到進(jìn)一步的驗(yàn)證.

圖2 銳鈦礦TiO2摻雜貴金屬前后能帶結(jié)構(gòu)圖((a)銳鈦礦TiO2;(b)Ru摻雜TiO2;(c)Pd摻雜TiO2;(d)Pt摻雜TiO2;(e)Ag摻雜TiO2;(f)Au摻雜TiO2)Fig.2 Band structures of undoped and noble metals doped anatase TiO2((a)anatase TiO2;(b)Ru doped TiO2;(c)Pd doped TiO2;(d)Pt doped TiO2;(e)Ag doped TiO2;(f)Au doped TiO2)
3.4 銳鈦礦TiO2摻雜前后電子態(tài)密度
銳鈦礦TiO2摻雜貴金屬前后電子總態(tài)密度和分態(tài)密度如圖3所示.從圖3(a)可以看出,單純銳鈦礦TiO2費(fèi)米能級附近的價(jià)帶主要由O的2p態(tài)構(gòu)成,導(dǎo)帶部分主要由Ti的3d態(tài)構(gòu)成,與文獻(xiàn)結(jié)果一致[20,24].圖3(b)-3(f)分別為Ru、Pd、Pt、Ag和Au摻雜TiO2后的總態(tài)密度和分態(tài)密度圖.從圖中可以看出,摻雜后Ti的3d態(tài)均出現(xiàn)了向低能方向的移動(dòng),與能帶圖中導(dǎo)帶下移相對應(yīng);O的2p態(tài)也在一定程度上從導(dǎo)帶向價(jià)帶移動(dòng).結(jié)合態(tài)密度圖可知,能帶中的雜質(zhì)能級主要為氧的2p態(tài)與貴金屬M(fèi)的d態(tài)(Ru、Pd和Ag為4d態(tài),Pt和Au為5d態(tài))雜化形成.Ru-4d、Ag-4d和Au-5d態(tài)與O-2p態(tài)雜化過程跨越了費(fèi)米能級,從而使其顯示出了金屬行為[16].而Pd的4d和Pt的5d態(tài)在摻雜的費(fèi)米能級處有一淵谷,從而導(dǎo)致了窄帶寬的出現(xiàn).

圖3 銳鈦礦TiO2摻雜貴金屬前后態(tài)密度圖((a)銳鈦礦TiO2;(b)Ru摻雜TiO2;(c)Pd摻雜TiO2;(d)Pt摻雜TiO2;(e)Ag摻雜TiO2;(f)Au摻雜TiO2)Fig.3 Densities of states of undoped and noble metals doped anatase TiO2((a)anatase TiO2;(b)Ru doped TiO2;(c)Pd doped TiO2;(d)Pt doped TiO2;(e)Ag doped TiO2;(f)Au doped TiO2)
3.5 銳鈦礦TiO2摻雜前后的光學(xué)性質(zhì)
在光學(xué)性質(zhì)計(jì)算中,采用“Scissors”算符對計(jì)算值進(jìn)行修正,修正值為1.077 eV,銳鈦礦TiO2摻雜貴金屬前后的吸收光譜如圖4所示.從圖中可以看出,與單純銳鈦礦TiO2相比,貴金屬摻雜后體系的吸收帶邊都有向可見光區(qū)擴(kuò)展的現(xiàn)象,體系在可見光區(qū)的吸收系數(shù)都有不同程度的增加.其中Ru、Ag和Au摻雜后體系吸收系數(shù)增加程度明顯大于Pd和Pt摻雜體系,這與能帶圖和態(tài)密度圖中分析的結(jié)果是對應(yīng)的.Ru、Ag和Au與氧形成的雜化能級跨越了費(fèi)米能級,大大減小了價(jià)帶電子躍遷所需的能量,從而更有利于提高體系在可見光區(qū)的吸收系數(shù),增大對可見光的利用率.Pd和Pt摻雜體系由于減小了禁帶寬度,也在一定程度上提高了在可見光區(qū)的吸收系數(shù).圖中Ag摻雜體系在可見光區(qū)出現(xiàn)了明顯的峰值,與Kumar等人[25]的實(shí)驗(yàn)結(jié)果基本一致.
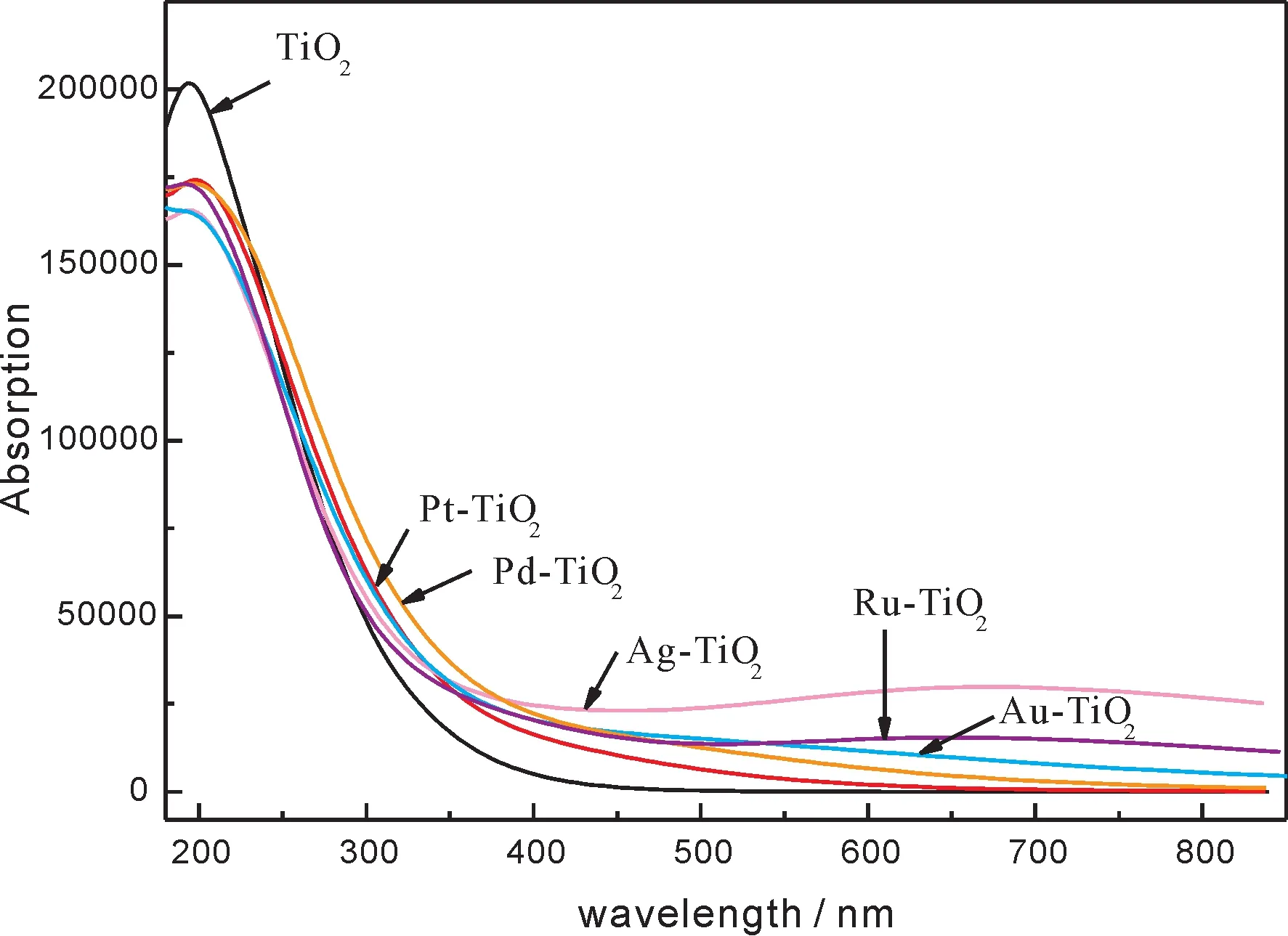
圖4 銳鈦礦TiO2摻雜貴金屬前后吸收光譜Fig.4 Absorption spectra of undoped and noble metals doped anatase TiO2
4 結(jié) 論
研究計(jì)算了貴金屬(Ru、Pd、Pt、Ag和Au)摻雜銳鈦礦TiO2的結(jié)構(gòu)性質(zhì)、電子特性和光學(xué)性質(zhì)變化.貴金屬摻雜后TiO2的晶格體積都出現(xiàn)了不同程度的增大,晶格出現(xiàn)了畸變.貴金屬摻雜TiO2的形成能在富氧條件下均為負(fù)值,表明摻雜是能夠?qū)崿F(xiàn)的,Ag、Au的形成能相較于其他貴金屬要小很多,說明其摻雜的熱力學(xué)穩(wěn)定性比其他貴金屬要好.Pd和Pt摻雜后體系的禁帶寬度減小,Ru、Ag和Au摻雜后體系表現(xiàn)出了金屬屬性,這主要是由于摻雜后形成了雜質(zhì)能級,雜質(zhì)能級主要為氧的2p態(tài)與貴金屬M(fèi)的d態(tài)(Ru、Pd和Ag為4d態(tài),Pt和Au為5d態(tài))雜化形成.Ru-4d、Ag-4d和Au-5d態(tài)與O-2p態(tài)雜化過程跨越了費(fèi)米能級,從而使其顯示出了金屬行為.而Pd的4d和Pt的5d態(tài)在摻雜的費(fèi)米能級處有一淵谷,從而導(dǎo)致了窄帶寬的出現(xiàn).貴金屬摻雜后體系的吸收帶邊都有向可見光區(qū)擴(kuò)展的現(xiàn)象,體系在可見光區(qū)的吸收系數(shù)都有不同程度的增加.
猜你喜歡
商品與質(zhì)量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛(wèi)生(2015年12期)2015-11-10 05:13:40
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
