瀝青質微觀聚集特征的分子動力學研究
2021-08-03 06:46:50黃世軍趙鳳蘭趙大鵬
油氣地質與采收率 2021年4期
關鍵詞:體系
王 鵬,黃世軍,趙鳳蘭,趙大鵬
(1.中國石油大學(北京)石油工程教育部重點實驗室,北京 102249;2.中國石油大學(北京)石油工程學院,北京 102249)
近幾十年來,瀝青質沉積問題給石油生產、運輸和煉油等領域帶來了許多工業挑戰[1]。瀝青質沉積不僅會造成儲層孔喉堵塞,還會導致井筒和生產設備腐蝕結垢,嚴重影響油田的正常生產[2-3]。因此,油田開發過程中瀝青質沉積的防治問題亟需解決。
瀝青質結垢的清除成本高昂,如何規避或抑制瀝青質沉積成為研究者們致力去尋找的解決方案。建立瀝青質沉積的熱力學預測模型是研究和預防瀝青質沉積的重要手段之一,而對瀝青質宏觀尺度熱力學行為準確建模的前提是需要了解其微觀尺度的聚集機理,這需要明確瀝青質分子聚集涉及的分子間作用力[4-5]。然而,由于瀝青質的分子結構和官能團尚未準確測定,瀝青質沒有精確的定義和統一的分子構型。瀝青質是石油中極性最強的重質組分,目前應用最廣泛的是根據溶解度將其定義為可溶于芳族溶劑(如甲苯)而又不溶于正構烷烴(如正庚烷)的化合物[6]。溶解度定義中提及的瀝青質一般特征不足以反映瀝青質分子聚集涉及的分子間相互作用,關鍵還是要獲取具有代表性的瀝青質分子構型。
隨著計算科學理論和計算機技術的進步,分子動力學方法得到廣泛的應用。分子動力學方法是在分子和原子尺度上描述體系結構與行為的計算機模擬方法,不僅可以用來預測物質的性質、材料微觀結構以及微觀作用機理,還可以解決時間和成本的問題,具有物理模擬實驗和理論計算不可比擬的優勢[7]。丁勇杰依據三組分分離法,選擇基于核磁共振分析和紅外分析的兩種瀝青質分子構型,采用分子動力學模擬方法,探究瀝青的化學結構特征[8];HEADEN 等開展經典原子分子動力學模擬研究,對比4種瀝青質分子構型、膠質分子構型及其混合物在甲苯或正庚烷中的聚集特征[9]。瀝青質分子結構對其聚集特征的影響較為顯著,且瀝青質分子結構多樣,每一個研究案例都需要單獨進行建模。因此,有必要建立一個具有代表性的瀝青質平均分子構型。為此,采用分子動力學方法,結合瀝青質分子單體代表性結構式、平均相對分子質量和分子基團特征,建立瀝青質平均分子構型,并將其應用到瀝青質模擬體系初始構型的構建,通過幾何構型優化和退火處理后,模擬瀝青質在地層環境中的運移過程,探究瀝青質分子性質和微觀聚集特征,進一步揭示瀝青質沉積的微觀作用機理。
1 模擬方法
1.1 瀝青質模擬體系的分子動力學實驗原理
分子動力學方法是基于經典力學原理,通過分子體系總勢能求解系統中所有原子核的牛頓運動方程數值解,分析原子核位置和速度隨時間的演化,來計算系統的動力學和熱力學性質[10]。量子力學方法從原子的電子結構和分子的運動出發,通過求解不同亞原子粒子的波函數來描述粒子行為,是最精確的分子勢能計算方法[7]。但即使是很小的系統,量子力學計算也非常昂貴,且超出了當前大多數計算機的計算能力。因此,研究者們建立了原子力場模型來近似計算分子勢能[10]。力場方法是在經典力學的理論基礎上,借助經驗和半經驗參數計算分子結構和能量的方法,它通過提供一組基于粒子相對位置的勢函數和力學參數來計算系統的勢能。
現有較為成熟的力場模型主要依據元素種類或原子所在官能團種類對分子中的各原子賦予不同的力場參數,計算分子內和分子間的相互作用,進而研究分子性質[11]。在力場模型中,化學鍵或所在官能團不同的同種元素會看作是不同種類的原子,這里的種類并非指真實的原子種類,而是力場模型中表征不同原子特征的分類,如烷烴上的碳、苯環中的碳和卟啉中的碳在力場模型中隸屬于不同原子,會賦予不同的力場參數。本文研究的瀝青質模擬體系,既包括有機大分子瀝青質單體,又包括水和二氧化碳等無機分子,對于這樣一個復雜的有機-無機復合系統,明確分子中原子的成鍵類型、官能團類型等結構特征,選擇合適的力場模型賦予相應的力場參數,是保證瀝青質模擬體系分子動力學模擬準確性的基礎。
1.2 模擬體系初始構型的建立
采用Accelrys公司研發的Materials Studio 軟件,建立瀝青質平均分子構型,并通過Amorphous Cell模塊組裝瀝青質模擬體系(圖1)。Amorphous Cell是一種分子動力學無定形組裝模塊,能建立復雜無定型系統中的代表性模型,以特定密度組裝多個分子,且各分子在模擬系統中的堆疊合理、仿真程度較高。以密度為1.2 g/cm3組裝4 個瀝青質分子、48個水分子和10 個CO2分子,模擬瀝青質在地層中的運移環境,以二氧化硅分子層代表地層孔隙骨架,模擬盒尺寸為26.60 ?×9.83 ?×12.99 ?,在各方向上均采用周期性邊界條件。
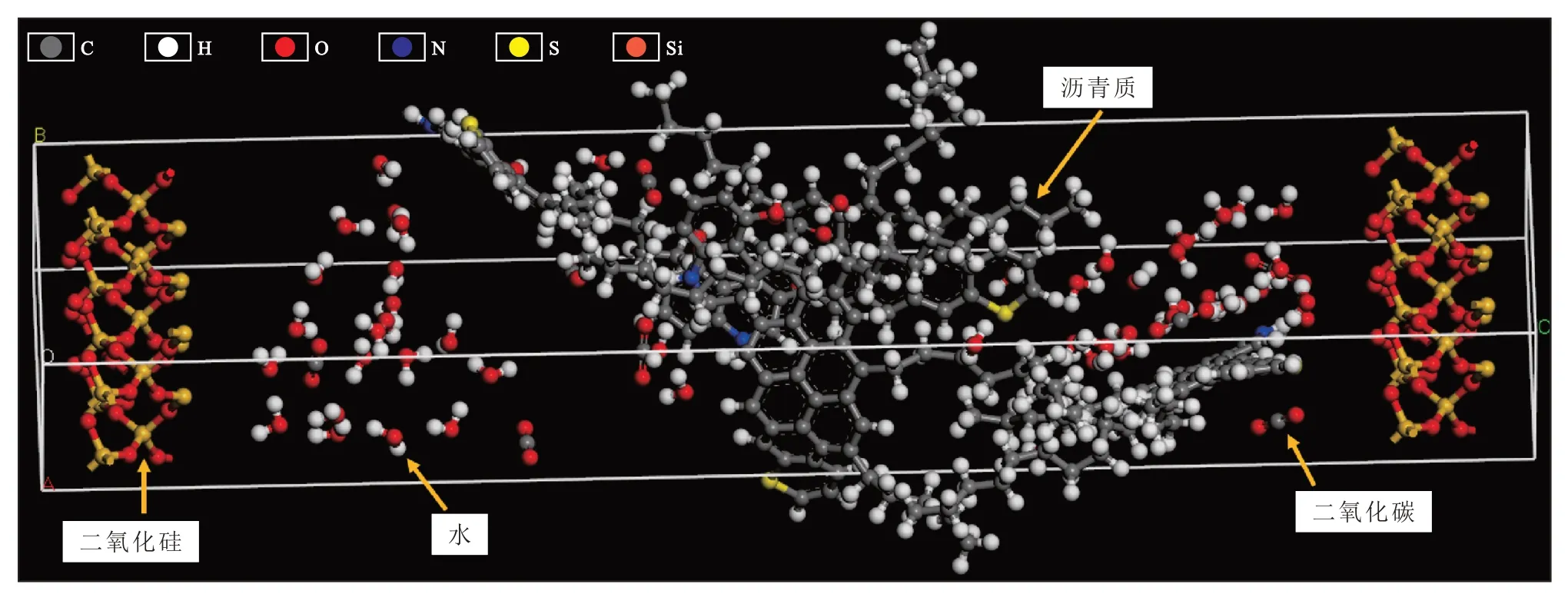
圖1 地層環境下瀝青質模擬體系的初始構型Fig.1 Initial configuration of asphaltene simulation system in formation environment
1.3 力場參數的確定
在進行分子動力學模擬實驗前,首先需要選擇合適的力場模型,并賦予相應的力場參數來計算粒子之間的相互作用,而力場參數的選擇對分子動力學模擬結果的準確性至關重要。不同的力場模型適用的模擬體系有所不同,如AMBER(Assisted Model Building with Energy Refinement)力場主要適用于模擬生物大分子,而CHARMM(Chemistry at Harvard Macromolecular Mechanics)力場可以較好地擬合小分子體系和溶劑化的大分子體系。因此,針對地層環境下瀝青質模擬體系的特點,從以下3 個方面考慮來確定力場參數。
瀝青質分子的自聚集行為 瀝青質作為原油中極性最強的組分,具有很強的自聚集傾向,不同濃度的瀝青質分子的聚集形態有所不同[4]。當質量分數較低(小于10-4)時,瀝青質以分散態的單分子形式存在;隨著體系中瀝青質質量分數的不斷增加,瀝青質分子開始發生聚集行為,聚集顆粒逐漸變大,依次形成納米聚集體(通常含有8~10個瀝青質分子)、納米簇和黏彈性網絡[12]。在選擇與瀝青質模擬體系相適應的力場模型時,首先要考慮的是能夠較好地擬合瀝青質這種有機大分子自聚集行為。
CVFF(Consistent Valence Force Field)力場可適用于計算各種多肽、蛋白質和大量的有機分子,在計算系統結構與結合能方面最為準確,可以較好地擬合瀝青質分子自聚集行為和瀝青質-膠質分子締合行為[13]。其勢能函數表達式為:

有機-無機復合體系 瀝青質模擬體系中除了有機大分子瀝青質,還包括水和二氧化碳等無機小分子,因此,需要選擇一種能兼顧有機-無機復合體系的力場模型。COMPASS(Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies)力場采用從頭計算與經驗方法相結合的方式計算力場參數,是第一個把有機分子體系的力場與無機分子體系的力場統一的力場模型[14]。其勢能函數公式為:
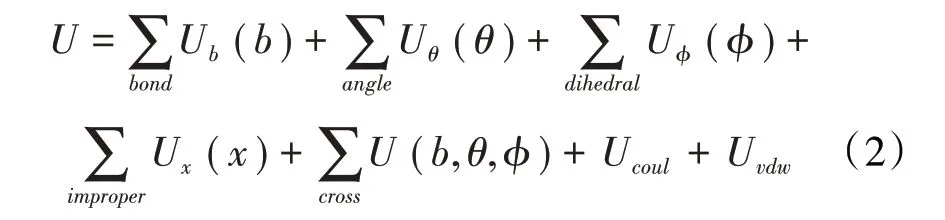
(2)式中右側前5 項為成鍵作用勢能,后2 項為非成鍵作用勢能,分別為靜電相互作用和范德華力,其計算公式分別為:

分子力場的遷移性問題 力場參數是通過大量擬合基分子的實驗數據得到的,若擬合的實驗數據是在常溫常壓條件下測定的,而模擬的瀝青質在地層中的運移環境往往是高溫高壓條件,此時應用這些力場參數時分子力學的準確性會降低[15]。Dreiding 力場是一種比較特殊的力場計算方法,可以計算一些缺乏實驗數據的新結構化合物,是典型的經驗力場模型。通過借助Dreiding 力場中的經驗參數進行輔助參考,有助于提高分子動力學模擬實驗結果的準確性。
1.4 瀝青質分子動力學模擬的實驗方法
瀝青質分子動力學模擬的實驗方法主要包括以下4 個步驟:①基于本文建立的瀝青質模擬體系初始構型,選擇合適的力場參數來計算系統中粒子間的相互作用。②采用最陡下降法調整偏離平衡位置較大的結構,再基于共軛梯度法對接近平衡狀態的結構進行優化,使系統內分子達到能量最小值和原子位置最佳分布,實現瀝青質模擬體系的幾何構型優化。③在高溫條件(303.15~503.15 K)下進行8個循環的退火處理,消除殘余應力,退火過程持續8 ps。④采用玻爾茲曼分布函數,隨機賦予系統中各分子運動的初始速度(圖2),使用Forcite 模塊在NVT 系綜下模擬瀝青質在地層巖石孔隙中的運移過程,模擬地層溫度設為328.15 K,控溫方法選擇Nosé-Hoover 算法,時間步長為1 fs,模擬時長為15 000 ps。

圖2 系統中各分子運動初始速度的玻爾茲曼分布Fig.2 Boltzmann distribution of initial velocity of each molecule in system
2 結果與討論
2.1 瀝青質平均分子構型的構建
瀝青質分子的主體由含有若干個芳環組成的片狀單元結構所構成,這些片狀單元結構的個體間結構差異較大,根據芳核特征可將瀝青質分子結構分為大陸型和群島型[16]。由不同地區原油中瀝青質的分子構型(圖3)可以看出,不同地區的瀝青質分子結構存在差異,如遼河和華北原油中的瀝青質分子的脂肪族支鏈較長,塔里木原油中的瀝青質分子則表現為標準的大陸型結構,而伊朗和俄羅斯原油中瀝青質分子的芳環數量較多[17]。雖然不同地區的瀝青質分子結構存在差異,但總體上看其結構特征也具有一般性,這為建立代表性的瀝青質平均分子構型提供了參考。
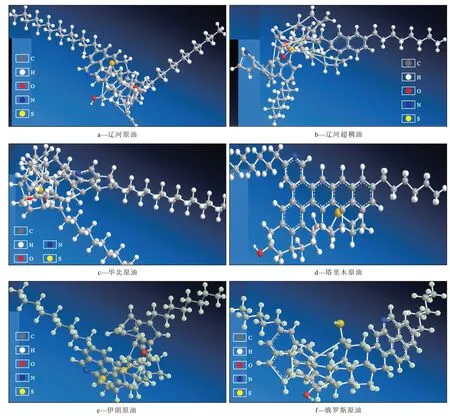
圖3 不同地區原油中的瀝青質分子構型Fig.3 Molecular configurations of asphaltenes in crude oil samples from different regions
目前,已知的大多數瀝青質分子單體結構包括1 個由4~10 個芳環組成的芳核和周圍長度為3~7個碳原子的脂肪族鏈,這為構建瀝青質平均分子構型的芳環數和支鏈長提供了參考依據[4]。ROGEL提出的一種基于沉淀法測定的委內瑞拉油樣瀝青質餾分的平均結構模型[18]被后續研究者沿用較多,其結構式具有一定的代表性,是本文瀝青質平均分子構型構建的基礎。而平均相對分子質量會影響瀝青質的密度和溶解度等物理性質,可以作為構建瀝青質平均分子構型的約束條件。隨著測定儀器精度的提高和測量方法的改進,瀝青質平均相對分子質量的測定值發生了幾個數量級的變化,從最初測定的103~106到現在普遍認可的750[19]。綜合多種瀝青質分子結構式、平均相對分子質量(750)和瀝青質分子基團結構的一般特征,構建出如圖4 所示的瀝青質平均分子構型,該分子構型的H/C 值為1.2,而瀝青質分子的H/C 值一般為1.0~1.5,因此其結構具有一定的代表性。

圖4 瀝青質平均分子構型Fig.4 Average molecular configuration of asphaltenes
2.2 瀝青質平均分子構型優化與退火處理
利用商業分子設計軟件構建分子構型的原理往往不是第一性原理,而是依靠分子內固有的化學鍵的鍵長和鍵角,但這些數據往往是通過大量實驗測得的平均數據,因此構建的初始分子構型往往不是目標化合物的穩定結構。為了得到更加真實的分子幾何構象,在分子動力學中通常采用能量極小化方法,即在分子間作用力處于最小的狀態下,調整分子幾何構象使分子鍵長和鍵角接近“自然”狀態,進而得到原子位置的最佳排布[10]。
從能量優化結果(圖5)可以看出,系統能量總體上隨著優化時間步數的增加而降低,但變化過程中存在明顯的轉折點。其原因是:由于勢能面具有波動性,數學上只能保證求得局部極小值,需要采用多種構象搜索方法結合使用,來盡可能地達到全局最小值[20]。本文首先采用最陡下降法,其原理是根據能量梯度負值進行一維搜索,是能量極小化初期最簡單、快捷的搜索算法,適用于分子結構偏離平衡位置較遠[10]。但隨著不斷逼近極小值,其搜索方向總是垂直于上一步的特性使得搜索到的勢能值總是在極小值附近震蕩而無法收斂,因而可以看到優化時間步數為0~380時能量衰減趨勢緩慢且波動頻繁,此時需要采用更加精細的共軛梯度算法,通過不斷迭代,快速逼近系統能量的全局最小值,該過程對應于優化時間步數為380~500時系統能量衰減速度顯著增加,直到達到最低勢能構象,即最穩定的構象。
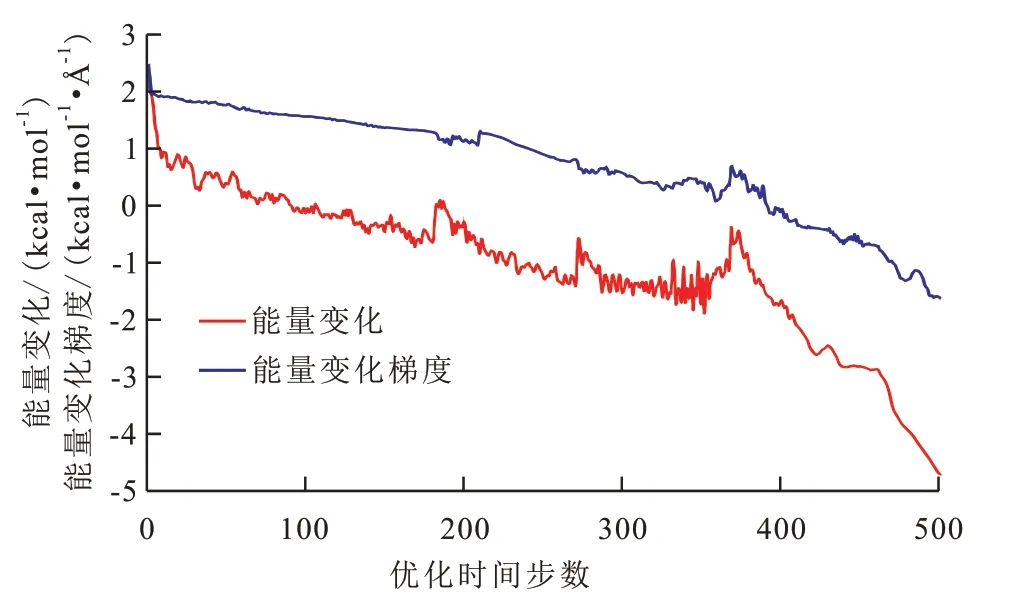
圖5 瀝青質模擬體系初始構型能量優化Fig.5 Energy optimization of initial configuration of asphaltene simulation system
對瀝青質模擬體系初始構型進行幾何構型優化后,完成系統溫度從303.15 K 到503.15 K 的8 個循環退火處理,退火過程中系統的各種能量變化如圖6 所示。由于退火過程中系統溫度波動,總動能和總勢能的變化也存在一定波動,但相比于初始構型,隨著系統溫度升高,分子熱運動加劇,系統總動能和總勢能增加。隨著分子間距增加,分子間的范德華力相互作用減弱,靜電相互作用將占據系統非成鍵作用中的主導地位。在高溫條件下,瀝青質分子內各原子熱運動加劇,能夠充分暴露其內部結構,隨后通過退火處理,系統溫度緩緩降低,在每個溫度都達到結構的平衡態,如此循環往復退火過程,消除殘余應力,使瀝青質分子結構達到能量基態。
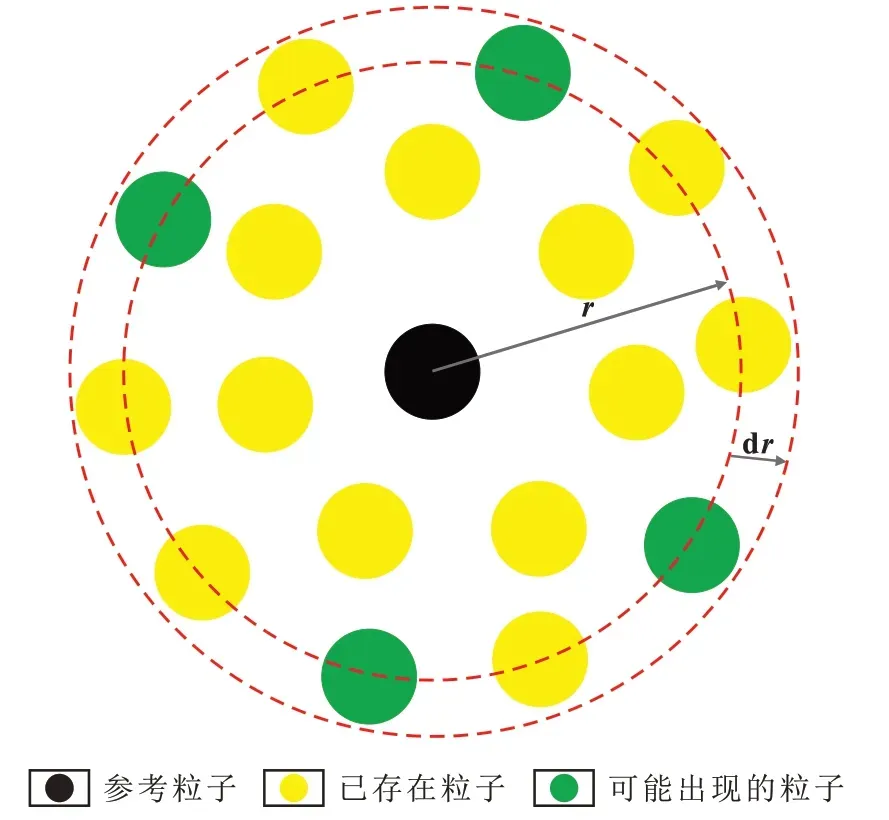
圖6 退火過程中系統能量變化Fig.6 Energy change during annealing
2.3 瀝青質分子構型的徑向分布特性
徑向分布函數是表征粒子微觀結構特征的物理量,其物理意義為任意指定的中心粒子周圍距離為r處出現其他粒子的概率,如圖7所示。它反映了凝聚態物質粒子聚集的徑向有序性,其特點為“以宏表微”,即只需測定瀝青質的宏觀數值就可以得到其微觀細節[21-23]。

圖7 徑向分布函數概率分布示意Fig.7 Probability distribution of radial distribution function
徑向分布函數可以理解為系統的局部密度與平均體密度的比值,其計算式[23]為:

分子動力學模擬獲得的系統各種統計學平均性質,主要通過統計分析函數來計算軌跡文件中關于各種性質的數據并繪制分布圖,如總能量、總勢能、總動能、速度和密度等隨時間的變化。g(r)同樣也可以從軌跡數據文件中獲得,其實質就是原子間矢量長度的球型平均分布[24]。對非晶體而言,當r值很大時,g(r)的函數值趨近于1,表明距離參考粒子為r的位置的粒子分布無規律性;而當r值相對較小時,g(r)的函數值會在小范圍內出現幾個波峰(極大值),即這些位置處的局部密度與平均體密度的比值較大,說明波峰位置處出現粒子的概率要明顯高于其他位置。總體上,非晶體的徑向分布特征表現為近程有序、遠程無序[25]。
從瀝青質平均分子構型的徑向分布函數(圖8)可以看出,g(r)總體上呈現近程波動劇烈且分散、遠程波動平緩且密集的特征。當r<0.25 nm 時,g(r)表現出較強的規律性,分別在0.11,0.14,0.17 和0.22 nm等處出現波峰,這些波峰表明從參考粒子到該位置處出現粒子的概率要遠高于其他位置,主峰位置一般標志著聚集結構的出現,這可能與瀝青質分子的稠環結構有關;而當r>0.5 nm時,g(r)的函數值趨近于常數1,說明粒子的分布沒有規律,呈無序狀態。瀝青質是典型的非晶體,在成鍵作用下分子內各原子呈有序排列狀態,表現為近程有序;隨著r值增大,瀝青質的分子聚集狀態方位會對瀝青質的出現頻率產生影響,在常見的方位配置中,具有非面對面配置方式的瀝青質分子徑向分布規律性較差,其出現頻率無規則,分子堆積更松散,因此遠程表現為無序狀態。

圖8 瀝青質平均分子構型的徑向分布函數Fig.8 Radial distribution function of average molecular configuration of asphaltenes
2.4 瀝青質在地層巖石中的運移模擬
瀝青質在地層環境運移過程中,系統內各分子運動的初始速度由玻爾茲曼分布函數隨機產生,模擬過程中系統溫度在設定值328.15 K上下波動。從瀝青質模擬體系在地層中的運移終態(圖9)可以看出:大量水分子向模擬盒的兩端運移,并最終傾向于吸附到二氧化硅分子層表面,這驗證了二氧化硅的強親水性;而二氧化碳分子在整個分子動力學模擬過程中位移量較小,這可能與其是非極性分子有關,且目前文獻中也尚未有記載關于從分子間的相互作用的角度去解釋二氧化碳對瀝青質沉積的直接影響;瀝青質分子中的芳核主體以及部分脂肪族鏈均不同程度的向二氧化硅分子層表面靠近,并傾向于逐漸被吸附到二氧化硅分子層表面,瀝青質分子單體或聚集體在二氧化硅表面的吸附造成孔隙內壁的非均質性會改變接觸角,是導致孔隙潤濕性改變最可能的原因之一,這一點在ANDREW 等[26]和LI 等[27]的實驗研究中都得到了驗證。巖石表面的潤濕性由親水性向親油性轉變,是瀝青質沉積導致原油采收率降低的重要機理之一。
此外,從圖9 中亦可觀察到瀝青質分子間的聚集狀態,聚集的驅動力來自于瀝青質分子中芳核的π-π 堆積作用,而脂肪族支鏈引起的位阻則是瀝青質分子聚集的阻力。π-π 堆積作用是由于芳香族環的負π 電子云和正σ框架之間的靜電吸引而產生的,常見的堆積方式包括面對面或平行、錯位堆疊或平行錯位、以及邊對面或T形[4]。其中,圖10中所示的堆積方式為面對面堆積,是最穩定的苯二聚體構型,它能夠最大程度地減少負π 電子云之間的相互排斥作用。瀝青質分子中芳核發生的π-π 堆積作用引發了瀝青質分子聚集,最終導致瀝青質絮凝并從原油中沉積出來,堵塞巖石孔喉并導致原油采收率降低。
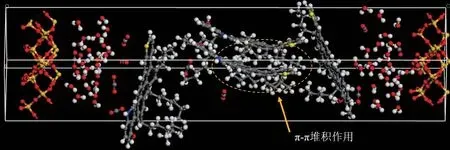
圖9 瀝青質模擬體系運移終態Fig.9 Final migration state of asphaltene simulation system

圖10 瀝青質分子二聚體構型示意Fig.10 Configuration of asphaltene molecular dimer
3 結論
基于分子動力學方法,結合多種瀝青質分子結構式、平均相對分子質量和瀝青質分子基團結構的一般特征,建立了瀝青質平均分子構型。該構型具有一定的代表性,并用于構建瀝青質模擬體系初始構型來模擬瀝青質在地層巖石中的運移。
瀝青質作為典型的非晶體,其徑向分布特征總體表現為近程有序、遠程無序。當周圍概率粒子到參考粒子的距離較小時,在成鍵作用下分子內各原子呈有序排列狀態;隨著周圍概率粒子到參考粒子的距離增大,具有非面對面配置方式的瀝青質分子徑向分布規律性較差,分子堆積更松散。退火處理過程中系統的各種能量變化表明,退火優化可使分子鏈充分松弛,有利于模擬該分子結構的能量初態。隨著分子間距增加,分子間的范德華力相互作用減弱,靜電相互作用將占據系統非成鍵作用中的主導地位。
從瀝青質模擬體系的運移終態可以看出,瀝青質分子的芳核主體及部分脂肪族鏈傾向于逐漸被吸附到二氧化硅分子層表面,這會使巖石表面的潤濕性由親水性向親油性轉變;瀝青質芳核的π-π 堆積作用是引發瀝青質分子聚集的驅動力,從而導致瀝青質絮凝并從原油中沉積出來。潤濕性反轉和巖石孔喉堵塞是瀝青質沉積造成原油采收率降低的重要機理,明確瀝青質分子的微觀聚集特征,能夠有效預防原油開采過程中潛在的儲層傷害風險,尤其是在注氣提高采收率技術應用過程中。
符號解釋
a,Dr——鍵伸縮能計算參數;
Aij——范德華相互作用參數,代表原子間的排斥作用;
b,b0——分子的鍵長和平衡鍵長,nm;
Bij——范德華相互作用參數,代表原子間的吸引作用;
dN——球形殼內的粒子數目,個;
dr——球形殼的厚度,nm;
g(r)——徑向分布函數;
i,j——體系中任意兩個不同原子;
kθ——鍵角彎曲能計算參數;
K?——鍵扭轉能計算參數;
kx——鍵角面外彎曲能計算參數;
n——鍵扭轉能、鍵角面外彎曲能參數;
qi——原子i的電荷量,C;
qj——原子j的電荷量,C;
r——周圍概率粒子到參考粒子的距離,nm;
rij——原子i與原子j之間的相對距離,nm;
——原子i與原子j間范德華勢能為0時的距離,nm;
U——總勢能,kJ/mol;
Ub——鍵伸縮能,kJ/mol;
Ucross——耦合能,kJ/mol;
Ux——鍵角面外彎曲能,kJ/mol;
Uθ——鍵角彎曲能,kJ/mol;
U?——鍵扭轉能,kJ/mol;
Ucoul——靜電勢能,kJ/mol;
Uvdw——范德華勢能,kJ/mol;
x,x0——分子的離平面振動角度、平均離平面振動角度;
θ,θ0——分子的鍵角、平衡鍵角;
?,?0——分子的二面角、平衡二面角;
ρ——密度,g/cm3;
ε0——介電常數;
εij——勢阱深度。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
