1,7?二氮雜15冠5醚對鑭(Ⅲ)的配位識別機理研究①
2021-07-12 06:51:34吳江華肖松文楊天足張杜超夏大廈萬洪強
礦冶工程 2021年3期
吳江華,肖松文,楊天足,張杜超,夏大廈,萬洪強
(1.中南大學 冶金與環境學院,湖南 長沙 410083;2.長沙礦冶研究院有限責任公司,湖南 長沙 410012;3.杭州研趣信息技術有限公司,浙江 杭州 310012)
基于超分子化學發展起來的分子識別技術,可針對特定的目標離子從離子半徑、配位特征、空間結構等多方面來設計合成配體分子[1],是一種新型高效選擇性分離技術。大環冠醚是人們研究最早、最透徹的一類陽離子識別主體化合物,被廣泛應用于分離萃取、藥物合成、催化反應等領域[2]。冠醚中的[—CH2—CH2—O—]基元,使其具有可通過離子?偶極相互作用與金屬陽離子成鍵的空腔,可根據空腔尺寸大小選擇性地與金屬陽離子結合,且金屬陽離子直徑大小與冠醚的腔穴尺寸越匹配,離子鍵合能力越強,所生成的配合物越穩定[2]。美國已有采用分子識別技術從溶液中分離稀土的應用實例,如IBC公司將冠醚負載于硅樹脂上制備出的SuperLig??188樹脂,可從阿拉斯加Bokan Dotson?Rigan重稀土礦提取液中成功分離出純度為99.999%的Nd和Dy[3-4]。截止到目前國內尚無采用分子識別技術富集分離稀土的相關報道,也未見硫酸體系中冠醚與稀土離子結合配位的相關機理研究。
1 實驗原理與方法
1.1 實驗原理
前期研究發現,氮雜冠醚可對輕稀土La3+表現出較強的選擇識別能力,但二者之間以何種方式結合配位尚不清晰。氮雜冠醚屬于中性含氮氧配體,原則上與稀土離子之間主要以氧合配位作用相結合。但由于稀土離子的配位數不固定(3~15不等),所形成的配合物結構與組成受中心稀土離子尺寸、配體結構、平衡陰離子等多重因素的影響[5]。
本文模擬南方離子吸附型稀土礦原地浸出液的溶液環境,基于量子化學計算和實驗測定相結合的手段來研究硫酸體系中氮雜冠醚萃取分離La(Ⅲ)的內在機理。基于DFT模擬計算構建稀土冠醚配合物的空間結構,初步確定配合物的化學組成;借助紅外光譜表征萃取前后有機物中極性官能團的結構變化,確定化學鍵結合方式;以氮雜冠醚為萃取劑、四氯甲烷為稀釋劑配置萃取油相,以硫酸鑭溶液為萃取水相,研究水相中離子濃度變化對La3+萃取分配比的影響,確定配合物中官能團的化學計量,從而揭示硫酸體系中氮雜冠醚與La3+配位識別的機理。
1.2 實驗方法
1.2.1 模擬計算分析
基于Gaussion進行建模計算[6],并采用Multiwfn 3.6對稀土?氮雜冠醚配合物分子結構進行模擬分析與幾何優化[7]。
1.2.2 萃取實驗
①硫酸稀土溶液的配置:氧化鑭加硫酸溶解后配置成0.000 5 mol/L的溶液,加稀硫酸調節水相pH=2.0,并添加硫酸鈉改變水相中游離SO42-濃度與體系離子強度。②有機相的配置:1,7?二氮雜?15冠5醚(2N?15C5)溶于四氯化碳配置成一系列不同濃度的萃取有機相。③萃取分配比的測定:萃取條件為25±1℃、相比1∶1、混相時間30 min、靜置分相時間60 min,分別考察水相中冠醚濃度及SO42-濃度對La3+萃取分配比的影響。
1.2.3 分析與表征
水相中La3+和SO42-濃度采用ICP?MS進行分析,負載La3+前后的有機相采用紅外光譜進行表征。
1.3 實驗試劑與儀器設備
實驗所用試劑及主要儀器設備分別如表1和表2所示。
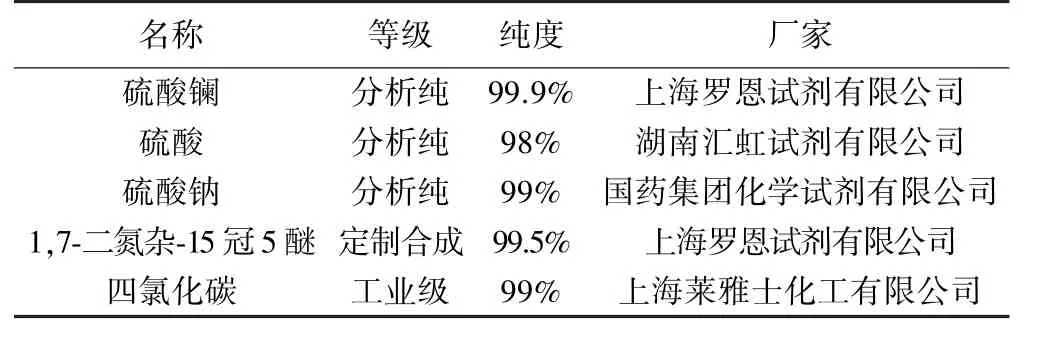
表1 實驗試劑明細

表2 主要儀器與設備
2 結果與討論
2.1 量子化學計算
基于常規腔穴尺寸效應機理,金屬離子直徑與冠醚環腔尺寸越接近,二者配位結合趨勢越大。La3+直徑0.212 nm,15冠5醚(15C5)環腔直徑0.208~0.243 nm;2個氮原子取代環上氧原子后,由于C—N鍵長與鍵角的改變,二氮雜15冠5醚(2N?15C5)直徑稍有增大,達到0.242~0.263 nm,如圖1所示。
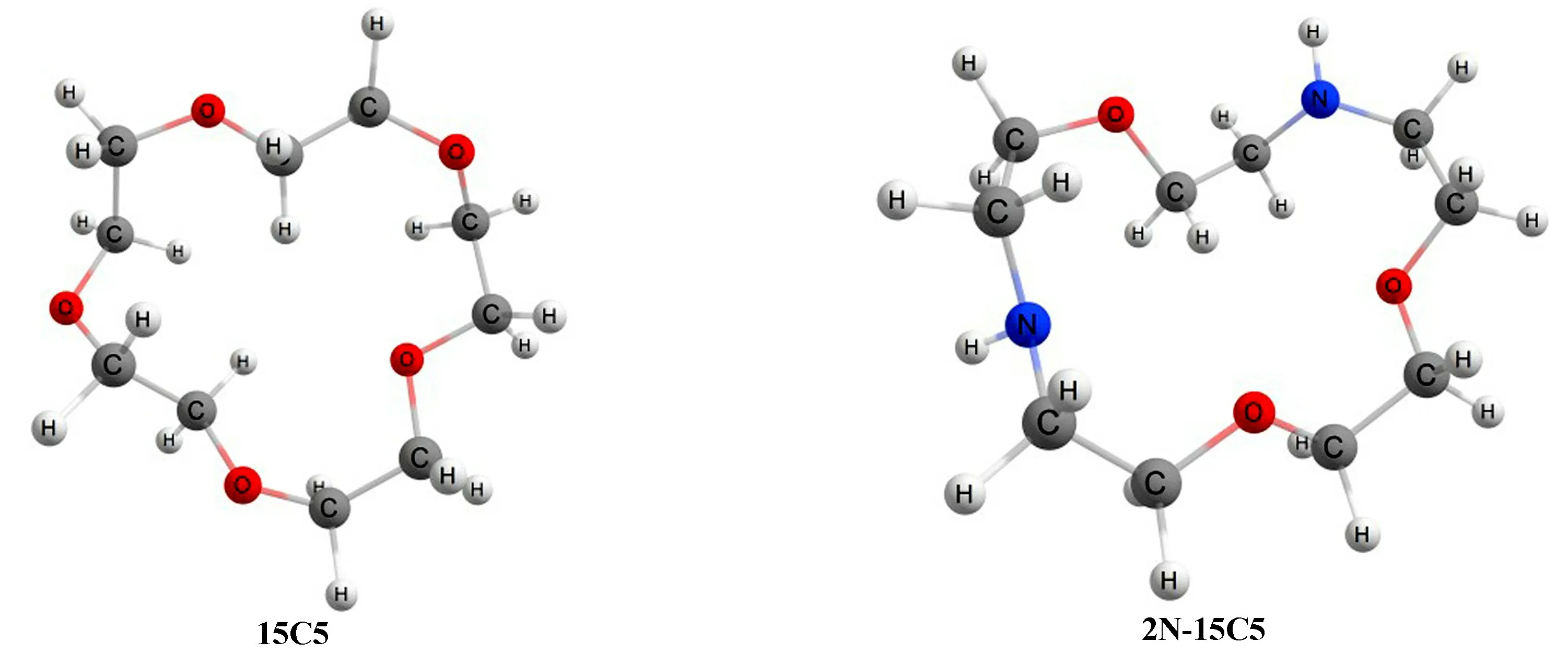
圖1 15C5與2N?15C5空間結構圖
基于B3LYP/6?311G(d)水平,模擬酸性水溶液環境,按照能量最低原理,分別以8配位和9配位構建La3+?2N?15C5配合物的配離子模型,結果顯示:8配位、La∶2N?15C5∶H2O=1∶1∶3的構型最穩定,如圖2所示。
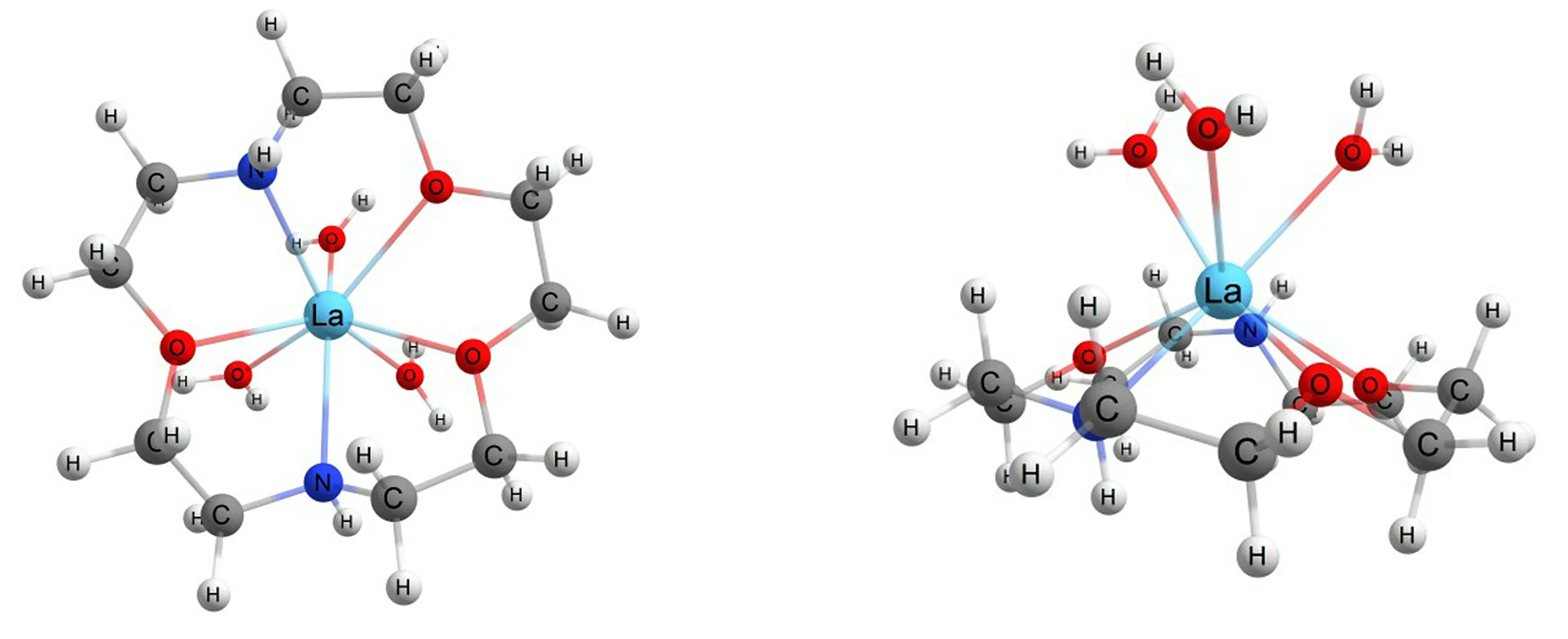
圖2 La(2N?15C5)(H2 O)33+配離子的空間結構圖
金屬離子La3+嵌在冠醚環腔中間,環上O原子和N原子與La3+成鍵,3個水分子中的O原子與La3+成鍵,且分布在冠醚環的一側。
采用Multiwfn 3.7進行波函數分析[7],得到以下結論:
1)配位結合能變。基于Gaussion計算模型,模擬水環境中La3+與冠醚的配位反應為:
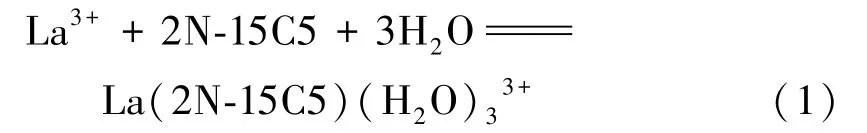
配位成鍵過程的配位結合能變為:
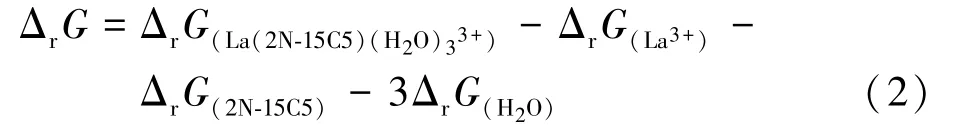
計算可得:ΔrG=-144.108 4 kJ/mol。
2)鍵長與鍵級。配離子中化學鍵的鍵長和鍵級數據能間接反映配離子的穩定性。一般來說,鍵長小于相互作用原子半徑之和,鍵級越大,化學鍵越穩定。配離子La(2N?15C5)(H2O)33+中各化學鍵的鍵長與鍵能數據如表3所示。

表3 La(2N?15C5)(H2 O)33+配離子的典型鍵長與鍵級數據
由表3可知,La3+與冠醚環上N、O原子和水分子上O原子之間確實存在化學鍵作用;La3+與冠醚環間作用力明顯強于其與水分子間的作用力,但又顯著弱于冠醚環上C—C、C—N鍵原子間作用力。說明該類作用力真實存在,但不屬于常規的共價鍵,而是更弱的原子間弱相互作用。
3)弱相互作用分析。化學體系中的弱相互作用主要包括范德華作用、π?π堆積作用、氫鍵、鹵鍵、配位鍵等,在弱相互作用分析中,sign(λ2)表示相互作用的類型,ρ表示相互作用的強度,且臨界點ρ是衡量相互作用強度的重要指標之一,其數值和鍵的強度(鍵級)存在正相關性。為進一步判斷La3+與N、O原子之間的作用力性質,對La(2N?15C5)(H2O)33+配離子進行弱相互作用(NCI)分析,結果如圖3所示。
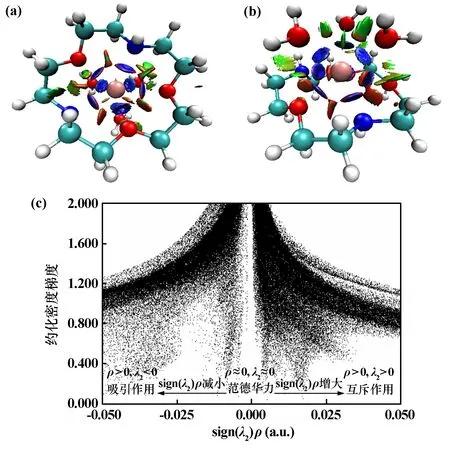
圖3 La(2N?15C5)(H2 O)33+的NCI圖與弱相互作用散點圖
弱相互作用的強度與相應區域的電子密度呈正相關[7]。一般來說,范德華力相互作用區域的電子密度很小,強空間斥力或相互吸引作用區域的電子密度很大。由圖3可知,冠醚環與La3+之間主要為吸引作用力(圖3(a)),體現為N、O原子與La3+之間的離子?偶極作用,同時相鄰N、O原子之間存在較弱的空間位阻斥力;3個水分子中的O原子與La3+之間主要為離子?偶極作用,水分子之間以及水分子與冠醚環之間存在范德華力(圖3(b))。
2.2 紅外光譜表征結果
為進一步探究氮雜冠醚與La3+的配位機理,對2N?15C5和負載La3+的氮雜冠醚(La?2N?15C5)進行紅外光譜表征,如圖4所示。
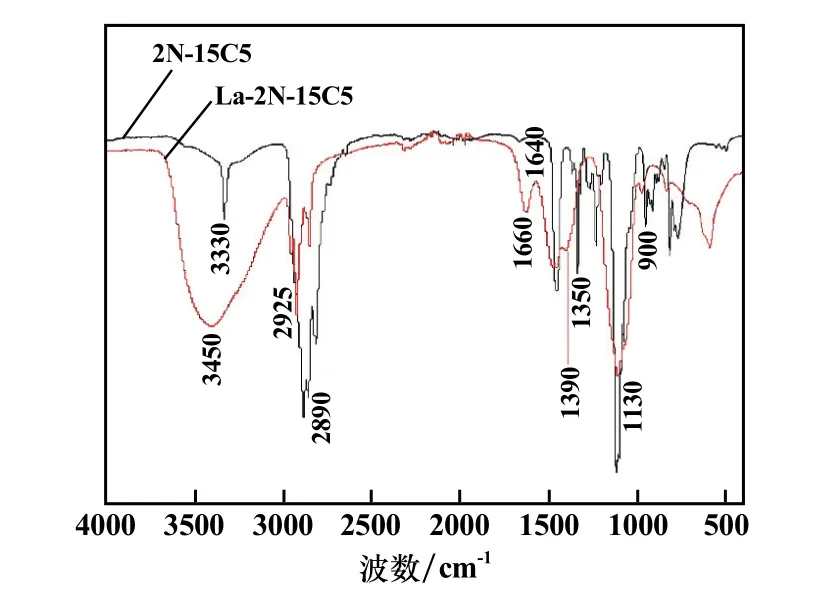
圖4 負載La3+前后的氮雜冠醚紅外光譜圖
在2N?15C5的紅外圖譜上,3 330 cm-1、1 640 cm-1、900 cm-1處的特征峰為N—H鍵,負載La3+后,有機物中的N—H特征峰被顯著弱化;1 250 cm-1處的C—N特征峰在配位反應后出現藍移,并與1 120 cm-1處的—C—O—C—特征峰疊加,峰強和峰寬均顯著增大。結合氮雜冠醚本身結構來看,該現象說明N、O原子均參與了配位反應,且屬于給電子配體,基于誘導效應致使相鄰—CH2基團的特征峰向高波數遷移,即2N?15C5紅外圖譜中2 890、1 470、1 350 cm-1處的特征峰,在負載有機物的圖譜中遷移到2 920、1 490、1 390 cm-1處,強度基本不變。
此外,在La?2N?15C5的紅外圖譜中,3 450 cm-1處出現了強且寬的H2O特征峰,且在1 660 cm-1處出現較強的氫鍵,證明水分子參與了配位過程。
2.3 配合物的結構組成
氮雜冠醚屬于中性含氮氧配體,與稀土離子之間主要以氧合配位作用相結合。根據前述模擬計算和紅外光譜測試結果可知,La3+、2N?15C5、H2O以氧合配位的形式組成1∶1∶3構型的配合物,但在萃取過程中SO42-是否參與配位成鍵還需深入探討。
基于徐光憲的配合理論[5],采用斜率法探究稀土與氮雜冠醚的配位絡合過程。冠醚?四氯化碳有機體系萃取La(Ⅲ)的反應包括冠醚在水相中的溶解、水相中的配合絡合反應、絡合物在有機相中的溶解3個步驟,各步驟對應的反應方程式如下:
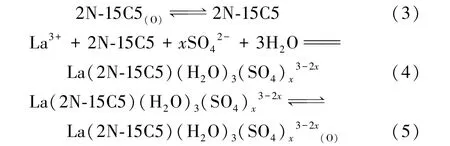
萃取平衡常數K計算式為:
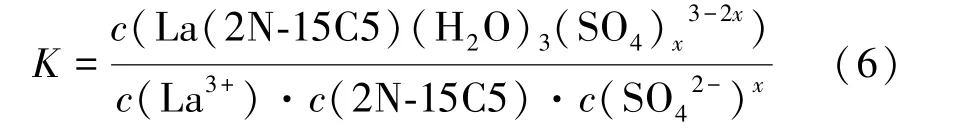
達到萃取平衡時,La3+在兩相中的分配比D為:

將式(7)帶入式(6),兩邊取對數可得:
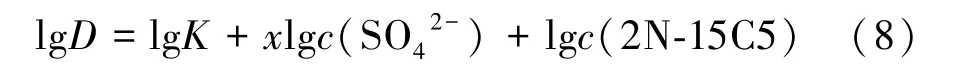
由式(8)可知,達到萃取平衡后lgK為定值,則lgD僅與水相SO42-濃度或冠醚濃度息息相關。由于水相中冠醚濃度主要取決于式(3)的溶解反應平衡,在實際萃取實驗過程中,有機相內冠醚含量是過量的,水相中2N?15C5濃度可視為某一未知的定值。因此,繪制lgD?lgc(SO42-)曲線即可根據曲線斜率獲得對應的x值。
中性絡合萃取中無H+參與反應,萃取前后水相pH值基本無變化,采用添加硫酸鈉來調整水相體系中的平衡陰離子SO42-濃度。在pH=2.0、相比O/A=1∶1條件下,lgD?lgc(SO42-)的相關關系如表4所示。
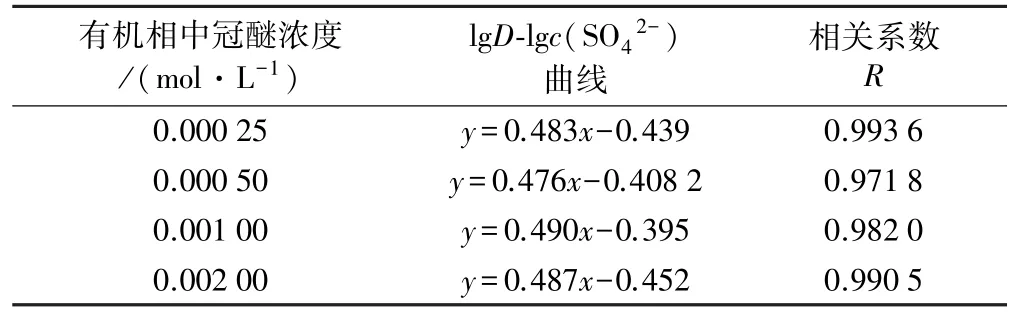
表4 lg D?lg c(SO42-)相關關系
上述擬合曲線的斜率集中在0.476~0.487范圍內,計算平均值為0.484。考慮到ICP?MS測試離子濃度所存在的誤差,可近似認為x=0.5。由此可確定硫酸體系中氮雜冠醚萃取識別La3+的反應式為:
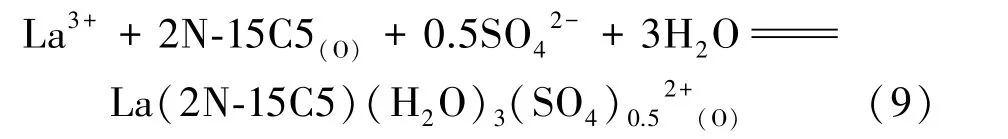
將萃取前后各物質的濃度值代入式(6),計算可得萃取平衡常數K=2.88×105。
3 結 論
1)模擬水環境中,La3+與氮雜冠醚形成La∶2N?15C5∶H2O=1∶1∶3的8配位結構,La3+落在冠醚空腔中,冠醚環上O、N原子與水分子中的O原子均通過離子偶極相互作用與La3+成鍵,配位過程的吉布斯自由能變為-144.108 4 kJ/mol。
2)在硫酸體系中,SO42-參與La3+與1,7?二氮雜?15冠5醚的配位反應過程,所得萃合物的結構式為La(2N?15C5)(H2O)3(SO4)0.52+,對應的萃取平衡常數K=2.88×105。
